Article rédigé par
SERGE ERLINGER, Médecin, professeur honoraire des Universités, ancien directeur de laboratoire INSERM
DOMINIQUE MORELLO, Chercheuse en biologie moléculaire, Directrice de Recherche au CNRS, retraitée. Membre de l’association Femmes & Sciences
Depuis la première lecture du génome humain au début des années 2 000, les progrès continus du séquençage de l’ADN permettent d’utiliser cette technique pour retracer notre histoire, étudier la biodiversité, suivre des épidémies, ou partir à la recherche de criminels. La génétique est devenue un outil incontournable. Quelques micro-traces d’ADN prélevées sur un défunt sont suffisantes pour nous renseigner sur les maladies dont il souffrait et les causes probables de sa mort. Ce récit s’inscrit dans une série de onze articles« De Beethoven à la Star’Ac : une enquête génétique et médicale », décrivant une dizaine de personnages illustres, morts ou encore vivants, chanteur, actrice, musicien, sportif, réalisatrice…, tous atteints d’une maladie héréditaire. Leurs maux sont décortiqués à l’aune de l’analyse de leur génome et des avancées médicales récentes.
Dans les années 2000, la Star Academy fait fureur sur TF1. Le 22 décembre 2004, Grégory Lemarchal remporte la quatrième saison avec 80% des suffrages du public. C’est la première fois qu’un garçon est finaliste ! Il est lancé. Mais 3 ans plus tard, Grégory doit définitivement quitter la scène. Il meurt à 23 ans des complications de la mucoviscidose. Son histoire singulière est l’occasion de découvrir une maladie génétique héréditaire due à un gène dont les mutations très fréquentes persistent pourtant depuis plusieurs millénaires. Mais pourquoi ?
Si Laurence Lemarchal avait tenu un journal, elle y aurait certainement relaté l’étonnement qui la saisissait lorsqu’elle embrassait son tout jeune fils Grégory : son baiser avait un goût salé qui n’augurait rien de bon. Elle a raison : lorsqu’il a 20 mois, le diagnostic de mucoviscidose est posé, la maladie s’installe, sans recours possible à un remède efficace, l’entraînant vers une mort inéluctable.
Tout commence au sous-sol du Babies Hospital
On rapporte qu’au XVe siècle déjà un proverbe circulait en Europe mettant en garde les parents : « Malheur à l’enfant qui laisse un goût salé lorsqu’on l’embrasse sur le front, car il est maudit et mourra bientôt« 1. Bien que connue depuis très longtemps, l’histoire contemporaine de la mucoviscidose commence en 1935 dans le sous-sol d’un laboratoire de pathologie du Babies Hospital à New York (l’hôpital pédiatrique du Columbia University Medical Center). A l’époque où seulement 5% des médecins en exercice aux États-Unis sont des femmes, Dorothy Hansine Andersen (1901-1963) est docteure en médecine de l’université Johns Hopkins et docteure en sciences médicales de l’université Columbia. Elle s’intéresse alors à une petite fille âgée de 3 ans qui présente une fibrose du pancréas et des lésions pulmonaires. Elle court les bibliothèques, accumule ses observations et publie en 1938 un article2 dans lequel elle décrit une cinquantaine de cas de patients présentant les caractéristiques de ce qu’elle appelle cystic fibrosis (CF, en français la fibrose kystique (FK) ou plus couramment mucoviscidose). Cette maladie, alors incurable, se caractérise principalement par des lésions du pancréas, des obstructions intestinales et des complications respiratoires, le tableau clinique et la sévérité de la maladie étant différents d’un individu à l’autre. De pionnière, elle devient experte.
Une sueur intrigante
Elle est rejointe en 1943 par le pédiatre Paul di Sant’Agnese (1914-2005) qui, pour soulager les jeunes patients de leurs infections, les traite à la pénicilline, un médicament alors rationné. Quand la pénicilline fut plus largement disponible, elle fit partie intégrante du traitement de la mucoviscidose.
Paul di Sant’Agnese remarque lors de la vague de chaleur à New York le 29 août 1948 la trace blanche que les enfants malades et assoiffés laissent sur leur verre d’eau. Il est ainsi le premier à s’intéresser à leur sueur. En comparant sa composition à celle d’enfants non malades, il observe une différence notable de concentration en sel (composé d’ ions sodium Na+ et chlorure Cl–), une donnée qu’il confirme par la suite sur un grand nombre d’enfants. Il établit alors une valeur seuil d’ions chlorure dans la sueur qui fut utilisée dès lors comme premier test de dépistage de la mucoviscidose3 (voir encadré).
Les années 50 virent aux États-Unis la cause de la mucoviscidose et son financement gagner du terrain avec la contribution notable de la pédiatre Wynne Sharples (1923-2008), fille d’un riche industriel et mère de deux enfants atteints de mucoviscidose. Elle crée en 1955 la National Cystic Fibrosis Foundation. En 1957, comme l’annonee un article du New York Times « La guerre contre la mucoviscidose est déclarée ».
Des efforts de nombreuses fondations, organismes de recherche et associations, dont celle de la famille de Grégory Lemarchal lancée en 2007 suite au décès de Grégory, ont fait considérablement avancer les recherches sur la mucoviscidose qui concerne environ une naissance sur 2500 en Europe et en Amérique du Nord4. Dans notre pays, on estime qu’environ 200 enfants atteints de cette maladie naissent chaque année, avec une grande disparité régionale (1/2500 dans le nord-ouest à 1/10000 dans le sud-est4).
Les progrès sont saisissants : il y a 90 ans, quand Dorothy Andersen établit son premier diagnostic, la plupart des enfants atteints de mucoviscidose ne survivaient pas au-delà de 5 ans. En 2005, l’espérance de vie d’un patient atteint de mucoviscidose est estimée en France à 47ans.
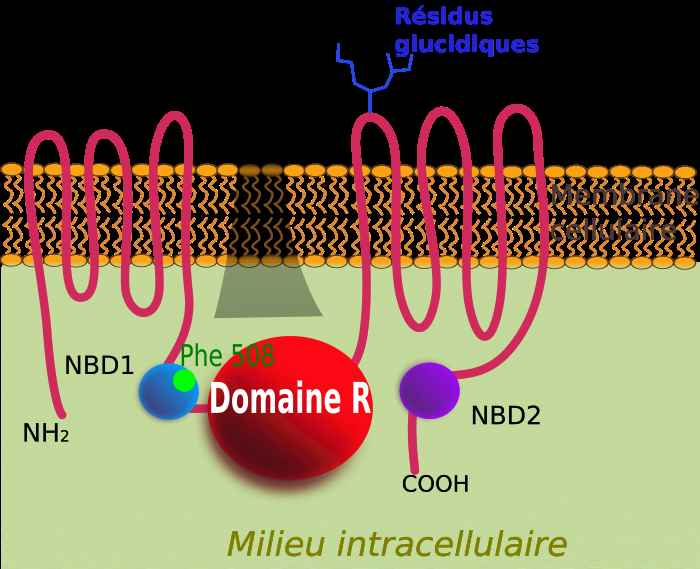
Après des années de traque, le gène responsable de la mucoviscidose est isolé en 1989 par une équipe canadienne dirigée par Lap-Chee Tsui (né en 1950). Sa découverte a révolutionné la recherche sur cette maladie monogénique (due au dysfonctionnement d’un seul gène), ses origines et les traitements5. Il s’agit du gène CFTR, localisé sur le chromosome 7 (voir encadré). Il code pour une protéine transmembranaire (traversant la membrane cellulaire) formant un canal à la surface des cellules épithéliales.
La protéine membranaire CFTR avec l’emplacement du site de la mutation Delta Phe508. Constituée de 5 domaines, elle se comporte comme un canal ionique laissant passer l’ion chlorure.bia University. https://www.cuimc.columbia.edu/news/legacy-changing-medicine-cystic-fibrosis. CC BY-SA 3,0 tonny, via wikimedia.
Cette molécule contrôle l’échange d’ions chlorure (Cl–) entre l’intérieur et l’extérieur des cellules. Elle joue ainsi un rôle fondamental dans la régulation du volume du liquide qui recouvre la surface des voies aériennes et dans la régulation de sa concentration en sel et de son pH. Chez les malades, ce canal ne fonctionne pas ou mal, entraînant des pertes de sueur riches en sel (NaCl) caractéristiques des baisers au goût salé. Il s’ensuit un déficit d’hydratation du mucus des voies respiratoires et digestives qui devient visqueux, d’où le nom de mucoviscidose, et, dans le poumon, la survenue d’infections bactériennes à répétition.
L’avantage hétérozygote
Depuis sa découverte, on dénombre près de 2 000 mutations indépendantes du gène6, la mutation appelée delF508 étant de loin la plus fréquente chez les personnes atteintes de mucoviscidose (voir encadré). Les hétérozygotes pour cette mutation (un seul de leurs deux chromosomes 7 porte le gène CFTR muté) ne sont pas malades : ils sont « porteurs sains ». Leur fréquence en Europe est estimée à 3-4%. En France, ce sont 2 millions de personnes porteuses saines d’une mutation responsable de la maladie. C’est énorme ! Étant donné que deux parents porteurs sains ont 25% de risque (1 chance sur 4) d’avoir un enfant homozygote donc atteint de mucoviscidose, comment se fait-il que la mutation n’ait pas été éliminée au cours de l’évolution ? Elle a très probablement été sélectionnée positivement au cours du temps. En remontant dans la préhistoire, les généticiens montrent que la mutation del508F est apparue en Europe au début de l’âge du Bronze (environ 3 000 ans avant notre ère) avec les éleveurs qui consommaient les produits laitiers de leur bétail7. Si l’hygiène laissait à désirer, des bactéries pouvaient s’y développer. Il semblerait que les hétérozygotes – qui ont donc 50% de canaux mutés – aient une meilleure résistance aux diarrhées dues aux contaminations par la toxine du choléra que les individus qui ne portent pas la mutation. C’est ce qu’on appelle l’« avantage hétérozygote ». On connait d’autres exemples d’un tel avantage, par exemple le « trait drépanocytaire » évoqué dans un autre article de cette série (lire « Drépanocytose : quand une mutation génétique devient un avantage évolutif… et touche un champion»). Ainsi, chez nos ancêtres, l’élevage aurait contribué à favoriser les porteurs sains de la mutation, qui se serait ainsi propagée à une fréquence élevée8.
Une révolution thérapeutique
La découverte du gène et de ses nombreuses mutations a permis de mettre au point un important arsenal thérapeutique pour une médecine de précision, transformant en à peine une génération une maladie pédiatrique en une maladie chronique de l’adulte, beaucoup de patients menant une vie quasi normale.
Citons en particulier l’avènement de petites molécule telles que les « correcteurs », améliorant le trafic intracellulaire de la protéine mal repliée, et les « potentiateurs », facilitant l’ouverture du canal dans la membrane9. Des combinaisons de ces différentes molécules sont actuellement en cours d’utilisation pour restaurer la fonction pulmonaire chez les porteurs de la mutation F508del, avec des effets souvent spectaculaires. Cependant, le coût d’une telle thérapie reste excessivement élevé (environ 300 000 €/an/patient), excluant un grand nombre de patients de ces traitements. Les recherches continuent toujours activement, en particulier celles concernant la thérapie génique qui consiste à introduire par nébulisation une copie fonctionnelle du gène, qui diffuse directement dans les poumons10.
A l’époque où Grégory était malade, peu d’approches thérapeutiques étaient disponibles, et seuls des traitements symptomatiques étaient proposés aux patients (antibiothérapie, anti-inflammatoires, kinésithérapie). Grégory était en attente de transplantation pulmonaire quand la maladie l’a emporté. Mais quel parcours remarquable cette star française, lauréat de l’émission Star Academy, a effectué avant de s’éteindre ! Depuis l’astéroïde 213637 qui porte son nom, à l’instigation de l’astronome amateur slovaque Stefan Kürti en 2020, Grégory Lemarchal doit contempler ces développements thérapeutiques avec envie !
Andersen, DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. American Journal of Diseases of Children 56, 344-99, 1938.
Le gène CFTR1 est localisé sur le bras long du chromosome 7. C’est un grand gène (215 kb) codant pour une grosse protéine (1480 acides aminés) qui forme un canal à la surface des cellules. Ce canal permet l’échange d’ions chlorure (Cl–) entre l’intérieur et l’extérieur des cellules. Depuis sa découverte, on dénombre près de 2 000 mutations du gène, la mutation appelée c.1521_1523del (F508del) étant la plus fréquente. Sur le gène, elle correspond à la suppression (ou délétion) de 3 paires de bases (CTT) en position 1521 à 1523. Sur la protéine, elle se traduit par la perte d’un acide aminé, la phénylalanine, en position 508 (p.F508del). Trois petites lettres manquantes au milieu d’une phrase qui en contient 4 440 et plus rien ne fonctionne ! Cette mutation dite sévère rend la protéine instable et le canal ne se ferme pas correctement. D’autres mutations touchent la synthèse de la protéine à différents stades de sa fabrication et affectent, suivant leur type, l’ouverture du canal, sa conductance ou sa disponibilité à la membrane.
pour Cystic Fibrosis Transmembrane Conductance Regulator, en français Régulateur de conductance transmembranaire de la fibrose kystique.
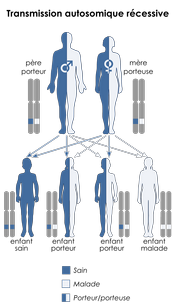
La mucoviscidose est une maladie récessive, ce qui veut dire que seul un enfant héritant d’une version mutée du gène de chacun de ses parents est atteint. A l’état homozygote (même mutation sur les deux allèles), la mutation p.F508del entraîne une insuffisance pancréatique et une atteinte pulmonaire précoce débutant dans l’enfance. La fonction pancréatique est en revanche préservée dans le cas où cette mutation sur l’un des allèles est associée à une autre, différente, sur l’autre allèle (ce qu’on nomme hétérozygote composite, voir épisode Beethoven, une enquête génétique tirée par les cheveux) et les infections bactériennes plus tardives. L’expression de la maladie est influencée par de nombreux facteurs (environnementaux, socio-économiques, et génétiques) de sorte que des patients ayant strictement la même mutation peuvent présenter de grandes différences dans la sévérité des symptômes et dans leur réponse à différents traitements.
Schéma de transmission autosomique récessive. CC BY-SA 3,0 Kashmiri, based on earlier work by Domaina.
Depuis 2002, en France, on dépiste systématiquement la mucoviscidose (et simultanément d’autres maladies génétiques rares) en prélevant une goutte de sang sur le talon du nouveau-né au troisième jour de la vie, goutte dans laquelle on dose une enzyme fabriquée par le pancréas (la trypsine immunoréactive ou TIR). Si sa valeur est élevée, on réalise le test sudoral mis au point par Sant’Agnese. Si le test est positif, on recherche sur le même prélèvement les principales mutations du gène CFTR responsables de la mucoviscidose classique.
Crédit image d’en tête : Kyrylo Ryzhov – adobe.stock.com / Double hélice ADN CC BY-SA 4.0 Joseluissc3, via Wikimedia
De Beethoven à la Star’Ac : une enquête génétique et médicale
Cet épisode concerne une anomalie génétique différente de celles décrites précédemment. En effet, elle n’est pas liée à un gène muté mais à un chromosome surnuméraire. Cette anomalie a porté plusieurs noms depuis sa première description par le médecin anglais John Langdon Down.
La dystrophie musculaire facio-scapulo-humérale, FSHD, est une maladie rare est une des myopathies héréditaires les plus fréquentes chez l’adulte. Mais à quoi est-elle due et quelles sont les pistes de recherche actuelles ?
Votre abonnement à la lettre d’information : Muséum de Toulouse
Le Muséum est ouvert !
Certaines zones limitées pourront toutefois rester temporairement fermées, et la capacité d’accueil est provisoirement réduite de moitié afin de préserver les collections.
Nous vous prions de nous excuser pour la gêne occasionnée et vous souhaitons une bonne visite.