Danse de St Guy ou maladie d’Huntington, une longue quête génétique
Modifié le :
Article rédigé par
SERGE ERLINGER, Médecin, professeur honoraire des Universités, ancien directeur de laboratoire INSERM.
DOMINIQUE MORELLO, Chercheuse en biologie moléculaire, Directrice de Recherche au CNRS, retraitée. Membre de l’association Femmes & Sciences.
Depuis la première lecture du génome humain au début des années 2 000, les progrès continus du séquençage de l’ADN permettent d’utiliser cette technique pour retracer notre histoire, étudier la biodiversité, suivre des épidémies, ou partir à la recherche de criminels. La génétique est devenue un outil incontournable. Quelques micro-traces d’ADN prélevées sur un défunt sont suffisantes pour nous renseigner sur les maladies dont il souffrait et les causes probables de sa mort. Ce récit s’inscrit dans une série de onze articles « De Beethoven à la Star’Ac : une enquête génétique et médicale », décrivant une dizaine de personnages illustres, morts ou encore vivants, chanteur, actrice, musicien, sportif, réalisatrice…, tous atteints d’une maladie héréditaire. Leurs maux sont décortiqués à l’aune de l’analyse de leur génome et des avancées médicales récentes.
Le célèbre chanteur folk Woody Guthrie, idole de Bob Dylan, mourut à 55 ans d’une bien étrange maladie héréditaire mêlant symptômes moteurs, cognitifs et psychiatriques. Face à un phénotype complexe et très variable, les scientifiques restèrent longtemps démunis. La famille Dexter, dont la maman « alias Mom », Leonor Sabin Wexler, était atteinte de la maladie, a beaucoup contribué à la recherche du gène coupable. Cette histoire est l’occasion de parler d’eugénisme, de diagnostics préimplantatoires et du temps parfois très long de la recherche.
Woody Guthrie (1912-1967) est un célèbre compositeur et chanteur folk. Texas, Californie, New York… ce « troubadour américain »1 compose à la guitare plus de 3 000 chansons folkloriques poétiques et protestataires. Il a inspiré toute une génération de jeunes musiciens, en particulier Bob Dylan, comme l’atteste la première chanson que Dylan composa lui-même en 1962 « A song for Woody Guthrie ». Quand Dylan arrive à New York, à peine âgé de 19 ans, pour rencontrer son idole, Woody Guthrie est déjà malade. Il séjourne à l’hôpital psychiatrique de Greystone Park (New Jersey). Il a été hospitalisé une première fois à sa demande sept ans plus tôt car, ne pouvant contrôler ses muscles, il craignait un trouble mental. Deux ans plus tard, il est arrêté errant dans les rues de Moristown (New Jersey). Après une nuit en prison, il est de nouveau transféré à l’hôpital de Greystone pour « schizophrénie paranoïde », où le personnel a du mal à croire que cet homme déboussolé a écrit d’innombrables chansons et le livre En route pour la gloire ! Quand Dylan le rencontre en 1961, il se déplace difficilement, son élocution est incertaine et son talent loin derrière lui. Mais Bob Dylan connaît son répertoire par cœur, un véritable juke-box, et rien ne met plus en joie Woody que d’entendre le jeune homme chanter ses propres mélodies. Dylan va très régulièrement voir Woody Guthrie à l’hôpital psychiatrique de Creedmoor (Queens) où il meurt à 55 ans. Bien qu’admis au milieu de malades mentaux, Woody n’en n’est pas un. Mais de quoi souffre-t-il donc ?
Une description précise de la maladie par Georges Huntington
Sa maladie a été décrite avec minutie en 1872 par le médecin américain Georges Huntington (1850-1916). Dans la famille Huntington, on est médecin de père en fils depuis plusieurs générations. Ainsi, Georges Huntington a-t-il été bercé par les histoires médicales contées par son grand-père Abel (1778-1858) et son père, Georges (1811-1881), tous deux médecins de famille à Long Island, près de New York. Un an après l’obtention de son diplôme de médecine, Georges Huntington fils fait, le 15 février 1872, à l’aube de ses 22 ans, une communication orale, devant l’Académie de Médecine des comtés de Meigs et Mason à Middleport dans l’Ohio, portant sur une forme de chorée inhabituelle, la chorée faisant référence aux tendances qu’ont les personnes atteintes à danser. Sa présentation est publiée quelques semaines plus tard dans le Medical and Surgical Reporter de Philadelphie2.
Il y décrit une « chorée héréditaire », une maladie qui s’éloigne de la chorée commune ou danse de Saint Guy par le fait qu’elle n’affecte pas les enfants mais concerne les adultes, rarement avant 30 ou 40 ans, qu’elle est héréditaire et incurable, contrairement à la danse de Saint Guy dont les enfants guérissent généralement à la puberté. Bien que les symptômes se ressemblent au début (contraction irrégulière et spasmodique de certains muscles), dans la chorée de Huntington, les symptômes s’amplifient avec l’âge, augmentant par degré. Chaque muscle du corps finit par être atteint, jusqu’à ce que « le malheureux qui souffre devienne une épave frémissante atteint de folie qui peut le pousser au suicide2 ».
Les premières descriptions de cette maladie remontent au milieu du 16e siècle en Norvège et au tout début du 17e en France et en Angleterre. Des colons anglais probablement atteints s’installent aux États-Unis dans les années 1630. Les noms de « désordre » et de danse de Saint Vitus sont relatés ; toutefois le caractère héréditaire n’est pas encore mentionné. Dans les années 1840, des médecins américains, anglais et norvégiens décrivent des personnes ayant des mouvements involontaires, des perturbations mentales et des parents présentant les mêmes symptômes. Ils nomment cette maladie « chorée héréditaire chronique ». C’est après la description minutieuse de Georges Huntington en 1872 qu’elle sera appelée maladie de Huntington (MH ou MD en anglais). L’intérêt pour cette forme de chorée est attesté par un nombre élevé de publications au début du 20e siècle.
Portrait de George Huntington par Irving Allison Watson, 1896. Wellcome Collection. Domaine public
C’est l’époque où les idées eugénistes, portées en particulier par le biologiste américain Charles B. Davenport (1866-1944), fleurissent. Il préconise, face à cette terrible maladie pour laquelle aucun remède n’est offert, « une stérilisation obligatoire et une restriction de l’immigration » afin d’empêcher les personnes atteintes et celles à risque d’entrer aux Etats-Unis !
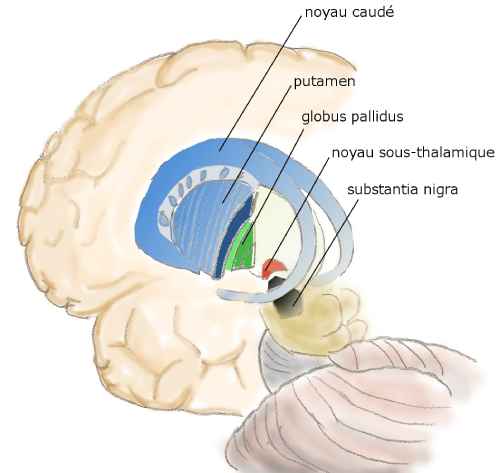
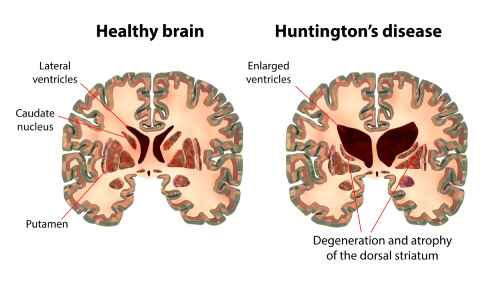
Si des traitements variés mais sans effets réels continuent d’être proposés, la neurophysiologie et la neurologie progressent au point qu’au début du 20e siècle la région du cerveau détruite par la maladie, le noyau caudé, est bien connue. Mais la cause de sa destruction reste mystérieuse jusqu’à la découverte du gène.
L’ex-femme de Woody Guthrie, Marjorie, danseuse professionnelle et mère de leurs 2 enfants Arlo et Nora, alors jeunes et à risque, fonde en 1967 le Comité de Combat contre la maladie de Huntington (CCHD). Il a pour but de rassembler les familles où sévit la maladie, de lever des fonds et faire pression auprès du Congrès pour stimuler la recherche. Marjorie propose, en outre, de nombreux services aux familles affectées et organise des campagnes d’information pour le public et les professionnels de santé. Elle organise en 1971 le fameux concert au Hollywood Bowl en hommage à Woody Guthrie, dont le profit va intégralement au CCHD.
Un an après la mort de Woody, une autre famille entre dans la danse. Elle jouera un rôle considérable dans la recherche et la découverte du gène responsable de la maladie. C’est la famille Wexler. Dans son livre paru en 1996 Mapping fate : a memory of family, risk and genetic research3, l’historienne et autrice américaine Alice Wexler relate comment elle découvre qu’elle et sa sœur sont à risque et décrit l’engagement de sa famille dans la recherche du gène responsable de la maladie.
Dans la famille Wexler – le père « Dad », Milton Wexler, la mère « Mom », Leonore Sabin, et les deux filles, Alice et sa jeune sœur Nancy – c’est le silence total sur la mort mystérieuse à 52 ans d’Abraham, le père de Mom, et sur la maladie qui emporte les 3 frères de Mom, Jess, Paul et Seymour, tous dans la force de l’âge. Ce silence est rompu en 1968 quand on diagnostique la MH chez Mom, alors âgée de 53 ans. Les deux filles, dans leur vingtaine, réalisent l’inconcevable et comprennent qu’elles ont une chance sur deux d’avoir hérité de la maladie. Le diagnostic de Mom incite leur père, psychiatre de renom, à créer la fondation HDF (Hereditary Disease Foundation) pour favoriser la recherche, récolter des fonds, organiser des rencontres, faciliter les échanges entre personnes qui n’ont à priori aucune raison de se côtoyer. Nancy, la sœur d’Alice, prend une part importante dans la gestion de cette fondation et joue un rôle décisif dans l’étude de la maladie. Les deux institutions, celle fondée par Marjorie et celle de Milton, sont très complémentaires ; l’une s’occupe plus spécifiquement des familles et des relations humaines, l’autre est le moteur d’une recherche pluridisciplinaire qui va au fil du temps concerner des neurologues, puis des généticiens, des biologistes moléculaires, des médecins, des virologistes, des immunologistes, des psychanalystes, des malades et leurs familles, et qui constituent un modèle de fonctionnement pour d’autres maladies génétiques héréditaires.
Mom mourût le 14 mai 1978, 10 ans après son diagnostic, 10 longues années de souffrance, d’anxiété, de dépression, de diminution inexorable physique et mentale. Alors que la MH affecte une population relativement limitée (actuellement 7 à 10 personnes pour 100 000, soit environ 30 000 aux USA et 6 000 en France), elle apparaît peu à peu comme un prototype pour la recherche médicale car elle altère une très large variété de fonctions.
Les premiers pas génétiques hésitants
Travailler sur cette maladie est une entreprise de très longue haleine. Ce n’est qu’en 1978 que le sujet se développe et que des scientifiques s’unissent pour trouver un marqueur génétique qui permettra de prédire qui est « destiné » à développer un jour la maladie et qui en sera exempt ; un test pour l’« HD-ness » qui servira aussi à comprendre l’anomalie biochimique à l’origine du mal, à éloigner la prévention reposant sur des pratiques eugénistes et offrira peut-être une réelle thérapie. Cinq ans plus tard, en 1983, ce marqueur est localisé sur le bras court du chromosome 4. La publication de cette découverte dans la prestigieuse revue anglaise Nature fait l’effet d’une bombe4. C’est la première fois que des généticiens mettent au point et utilisent une nouvelle technique de biologie moléculaire (la RFLP, pour Restriction Fragments Length Polymorphism, voir encadré RFLP), pour localiser sur un chromosome un gène responsable d’une maladie génétique héréditaire, alors qu’aucune piste n’était privilégiée. Et cette démarche ouvre la voie à l‘étude de gènes dont les mutations provoquent d’autres maladies héréditaires, comme la dystrophie musculaire de Duchenne (1986) et trois ans plus tard la mucoviscidose (voir l’épisode Baiser salé).
Mais pour la MH, le chemin est encore long avant la découverte du gène lui-même, fruit d’un véritable travail collaboratif impliquant une dizaine d’équipes, signant leurs publications d’un seul et même nom : le groupe de recherche collaborative sur la maladie de Huntington. Bravo ! Ce n’est pas bien courant en recherche. Et cette remarquable dynamique paye : la découverte du gène, nommé alors IT15, est annoncée le 24 février 1993 et publiée le 26 mars de la même année dans la célèbre revue scientifique américaine Cell5. La publication relate l’histoire de la découverte du gène. C’est la recherche la plus longue et la plus frustrante des annales de biologie moléculaire .
Le gène HTT
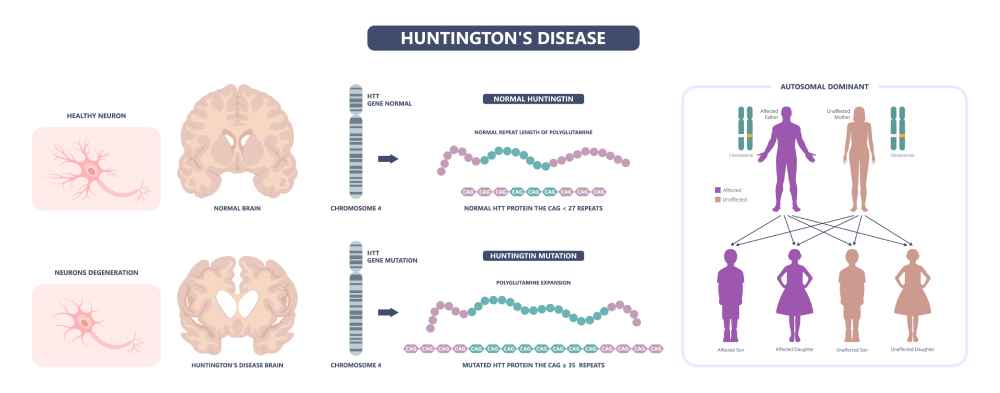
Le gène, appelé maintenant HTT, est très long, environ 50 fois plus que la taille moyenne d’un gène humain, et la protéine pour laquelle il code, la huntingtine (HTT), ne ressemble à rien de connu !
La MH est une maladie génétique, héréditaire, dont la transmission est dominante, c’est-à-dire qu’il suffit d’un gène muté reçu de l’un ou l’autre des parents pour en être atteint (voir encadré gène HTT et fonction de la protéine huntingtine). En d’autres termes, une personne dont l’un des parents est malade a une chance sur deux d’être affectée à son tour. Le porteur du gène défectueux développera la maladie inéluctablement à un moment de sa vie. Comme disent les généticiens, la pénétrance du gène est complète. C’est le cas de Woody Guthrie et de Mom qui ont hérité de leur mère ou de leur père, respectivement, un gène HTT muté.
La découverte du gène responsable de la MH permet enfin de prédire avec certitude qui développera ou non la maladie, un souhait formulé depuis longtemps par les généticiens, les professionnels de santé et certaines familles. Mais à quel prix ? Savoir ou non, avant que les symptômes n’apparaissent, qu’on va développer à coup sûr une maladie incurable, sans connaître précisément quand, constitue un dilemme cornélien pour la personne concernée et ses proches. Aujourd’hui, malgré la facilité des tests, une grande majorité des personnes à risque ne se fait pas tester (plus de 90% aux États-Unis et en Europe). Cependant, pour les couples dont un des membres est à risque et qui souhaitent connaître le statut génétique de leur futur bébé, il est possible d’effectuer un diagnostic préimplantatoire6 (voir encadré diagnostic prénatal et pré-implantation).
La maladie de Huntington est l’illustration par excellence du temps long de la recherche : l’année 2023 marque le 150è anniversaire de la description minutieuse qu’en fit Huntington, le 40è de la découverte d’un marqueur à quelques pas du gène, et le 30è anniversaire de la compréhension de la mutation génétique. En 150 ans, d’immenses progrès ont été réalisés, mais malgré des recherches prometteuses7 de grandes inconnues subsistent. Sensibiliser à cette maladie dégénérative pour promouvoir encore plus de recherches reste toujours d’actualité. Et vivre avec fougue le présent, comme le montre si magistralement Dimitri Poffé, récemment testé positif pour la MH, qui a entrepris un voyage de 18 000 km du Mexique à la Patagonie à vélo : « Ma vie ne sera peut-être pas longue, mais au moins elle sera courte et riche »8.
Dimitri Poffé parcours l’Amérique latine en vélo pour sensibiliser à la maladie de Huntington, fédérer une communauté et collecter des fonds. Crédit Projet Explore for Huntington.
Mapping fate: a memory of family, risk, and genetic research (1996, California Ed.). C’est de ce livre, non traduit, qu’est tirée la plupart des informations de cette nouvelle.
A polymorphic DNA marker genetically linked to Huntington’s disease. J.F. Gusell, NS Wexler, PM Conneally et al. Nature, 1983, 306, 234-8. doi: 10.1038/306234a0.
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s disease collaborative research group (no authors listed). Cell 72, 971-83, 1993.
Tabrizi, SJ et al. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nature Reviews Neurology 16, 529-546, 2020.
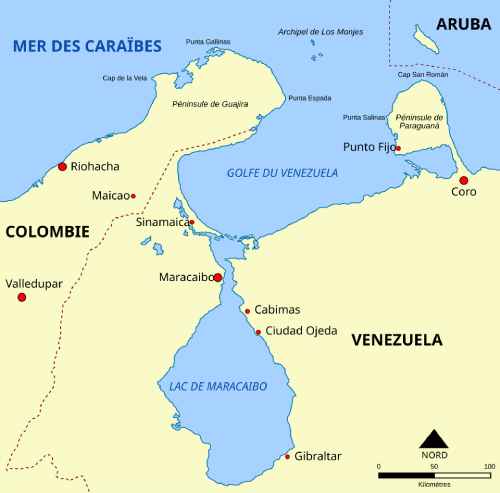
Partir à la recherche d’un gène quand on n’a aucune idée du chromosome sur lequel il se trouve n’est pas une mince affaire, surtout dans les années où la biologie moléculaire n’a pas encore pris son plein essor. La stratégie choisie alors, en 1983, pour localiser le gène dont la mutation est responsable de la maladie de Huntington (HD en anglais ou MH) est basée sur la technique dite de RFLP ou Restriction Fragment Length Polymorphismi. Le mot restriction se réfère à l’utilisation d’enzymes dites « de restriction », des enzymes d’origine bactérienne qui coupent l’ADN dans une séquence de nucléotide très précise. L’ADN à l’étude, ici l’ADN humain long de 3 milliards de nucléotides, est ainsi coupé en de très nombreux fragments appelés « fragments de restriction ».Pour leur recherche, une équipe de scientifiques menée par Nancy Wexler s’est rendue durant 3 ans, un mois par an, sur les rives du lac Maracaibo, au Venezuela, lac autour duquel résidait une communauté comportant de nombreux individus ravagés par la maladie de Huntington.
Localisation du lac de Maracaibo au Vénézuela CC by-sa 3.0 Kimdime69, via wikimedia
Pêcheur prenant un bain dans le lac de Maracaibo au Vénézuela. CCO 1.0 Wilfredor, via wikimedia.
L’équipe a recueilli une grande variété d’informations sur les liens de parenté entre les individus, leur état clinique (sain ou malade et à quel stade) et a prélevé sur chaque individu suffisamment de matériel (biopsie de peau et sang) pour en extraire de l’ADN et entreprendre une analyse RFLP. Après de longs mois d’effort, l’équipe a identifié un fragment de restriction caractéristique des malades. Des études complémentaires ont permis de le localiser sur le chromosome 41. Il a fallu attendre encore 10 ans avant que la localisation précise du gène huntingtine et sa séquence soient connues, et que la nature exacte de la mutation soit révélée !
Gusella, J.F et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature, 306, 234–238, 1983.
Pour les couples qui souhaitent un enfant et dont l’un des membres est ou pourrait être porteur d’une mutation responsable de la maladie de Huntington, deux types de diagnostics sont proposés : le diagnostic prénatal si l’un des parents est porteur du gène muté, et le diagnostic préimplantatoire lorsqu’au moins un des futurs parents est à risque de porter le gène responsable de la maladie, qu’il connaisse ou non son statut.
Le diagnostic prénatal consiste à rechercher chez le fœtus, soit par amniocentèse, soit par prélèvement d’un petit fragment du futur placenta, généralement entre la 11e et la 13e semaines d’aménorrhée, une mutation du gène huntingtine (HTT). Si le résultat est positif, les parents peuvent choisir d’interrompre la grossesse.
Le diagnostic préimplantatoire se fait dans le cas où les couples choisissent une fécondation in vitro (FIV). L’anomalie génétique est recherchée sur une des cellules des embryons obtenus par FIV, trois jours après la fécondation. Seuls les embryons exempts de la mutation sont transférés chez la mère pour une implantation dans l’utérus.
Le gène humain huntingtine (HTT) se trouve sur le chromosome 4. Dans sa version normale, on trouve en son début un triplet de nucléotides (CAG) répété entre 11 et 33 fois, en moyenne 20 fois. Mais chez les malades, ce triplet est répété 37 à 100 fois ou plus. Pour ceux qui présentent une répétition de 30 à ~ 40 on ne peut pas prédire s’ils développeront ou non la maladie. En revanche, quand le nombre de répétitions est supérieur à 40, la fonction du gène est altérée et la conformation de la protéine mutée est anormale. Il existe un test rapide pour détecter les patients avec un nombre de triplets élevé1. Plus la longueur de la répétition est longue, plus précoces seront les symptômes2. Les répétitions sont instables, leur nombre pouvant changer quand elles sont transmises d’un parent à son enfant. La transmission de cette maladie génétique héréditaire est dominante, c’est-à-dire qu’il suffit qu’un seul allèle muté soit présent pour que la maladie se développe.
Mutation, répétitions du triplet de nucléotides CAG et dominance de l’allèle muté dans la maladie de Huntington. D’après illustration Pepermpron, AdobeStock.com (ici version corrigée).
La protéine huntingtine est indispensable pour le développement embryonnaire et joue un rôle vital dans la neurogenèse. Elle est impliquée dans de nombreuses fonctions cellulaires. Les neurones qui expriment la protéine mutée meurent peu à peu et n’assurent plus les nombreuses fonctions qui leur incombent (motrices, oculomotrices, cognitives et limbiques, contrôle du comportement et des émotions). Les effets toxiques dus à la protéine mutée sont liés à deux phénomènes principaux : formation d’agrégats avec de nombreuses protéines, fragilité de la protéine mutée qui est coupée en fragments dont certains s’accumulent dans le noyau du neurone et provoquent la mort neuronale.
Comprendre l’importance et le fonctionnement du triplet CAG dans l’émergence de la maladie MH est capital, d’autant que d’autres maladies génétiques héréditaires sont aussi caractérisées par une extension de triplets. On en compte actuellement une vingtaine dont la maladie de Kennedy, le syndrome du X fragile ou l’ataxie de Friedreich.
Malbec, R. et al. µLAS: Sizing of expanded trinucleotide repeats with femtomolar sensitivity in less than 5 minutes. Scientific Reports, 9, 23, 2019.
Duyao, M. et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nature Genetics, 4, 387-392, 1993.
Quand votre mère, votre grand-mère, votre tante ont eu un cancer du sein ou de l’ovaire, quel est le risque que vous en ayez un vous-même ? L’analyse de l’ADN permet aujourd’hui de répondre à cette question. C’est ce qu’a fait Angelina Jolie.
Le chanteur Woody Guthrie mourut d’une étrange maladie héréditaire mêlant symptômes moteurs, cognitifs et psychiatriques. La famille Dexter a beaucoup contribué à la recherche du gène coupable. Cette histoire est l’occasion de parler d’eugénisme, de diagnostics préimplantatoires et du temps parfois très long de la recherche.
Votre abonnement à la lettre d’information : Muséum de Toulouse