En ce cinquième anniversaire du confinement en France, cet article propose une synthèse des données scientifiques et autres révélations postérieures à juillet 2021 sur l’origine et les répercussions du Covid 19, faisant suite aux précédents articles écrits avant cette date par les mêmes auteurs1-4. Officiellement, en cinq ans, 776,8 millions de cas ont été recensés dans le monde et 7 millions en sont mortes. D’après l’OMS, une estimation plus réaliste serait autour de vingt millions de morts. En France, on en dénombre au moins 168 000. De redoutable, le SARS-CoV-2 est devenu aussi banal que le virus de la grippe. Le virus s’est adapté et nous avec, à force d’infection et de vaccination. Actuellement, aucune preuve définitive ne peut, à elle seule, permettre de trancher entre l’hypothèse zoonotique (maladie transmise de l’animal à l’humain) ou la fuite accidentelle ou intentionnelle d’un virus manipulé en laboratoire. Cela n’empêche pas de disposer de nombreux éléments plaidant en faveur de l’une ou l’autre de ces hypothèses. Nous présentons ici les principales données disponibles qui permettront à chacun de se faire sa propre idée.
Premiers pas d’une propagation virale
Il y a cinq ans, le 30 janvier 2020, le directeur général de l’Organisation mondiale de la santé (OMS) Tedros Adhanom Ghebreyesus déclarait « une urgence de santé publique de portée internationale concernant l’épidémie mondiale du nouveau coronavirus », l’agent pathogène jusqu’alors inconnu qui avait tué en Chine 170 personnes et s’était déjà propagé dans dix-huit pays. La transmission interhumaine du virus était établie et l’on savait depuis peu qu’elle débutait avant l’apparition des symptômes. Mais les données nous parvenaient au compte-goutte du fait des précautions, de la censure et de la réduction au silence des lanceurs d’alerte chinois par leur gouvernement, à l’image du médecin ophtalmologiste Li Wenliang, qui a été l’un des premiers à alerter sur l’apparition d’un nouveau coronavirus (SARS-CoV-2) à Wuhan fin 2019 en partageant ses inquiétudes avec des collègues médecins sur l’émergence de cette maladie ressemblant au SRAS. Cependant, les autorités chinoises l’ont réprimandé, l’accusant de « propagation de rumeurs ». Il aurait ensuite contracté le virus et en serait décédé le 7 février 2020 à l’âge de 34 ans, suscitant une vague d’émotion et de colère en Chine et dans le monde entier. Son histoire est devenue emblématique du manque de transparence initial dans la gestion de la pandémie, à l’instar de l’ex-avocate Zhang-Zhan, libérée en mai 2024 après 4 ans d’emprisonnement, dont la parole reste muselée. Le confinement général avait été décrété le 23 janvier à Wuhan, la capitale de la province du Hubei hébergeant près de 12 millions d’habitants, dont le marché d’Huanan, d’où tout serait semble-il parti. Les scientifiques chinois détenaient depuis plusieurs jours la séquence du SARS-Cov2, le virus responsable de ce que l’OMS nommera Covid-19 le 11 février. Quelques pays prennent alors des mesures face à l’urgence, d’autres, comme les Etats-Unis et l’Italie, y sont indifférents. La France tarde ; les premiers morts sont décomptés et ce n’est que le 11 mars, jour où l’OMS déclare la pandémie, qu’elle réagit officiellement, avec la création du conseil scientifique Covid-19, suivie le 16 mars, le lendemain du premier tour des élections municipales, de la déclaration du président de la République Emmanuel Macron : « Nous sommes en guerre » et du mot d’ordre de confinement prononcé à 22h par Christophe Castaner, le ministre de l’intérieur d’alors. Bilan ce jour-là : 7 500 cas déclarés, 2 279 hospitalisations et 148 décès. Aucun traitement efficace n’est disponible, les tests et les masques manquent. Le premier ministre Edouard Philippe déclare le 13 mars « Porter un masque en population générale, ça ne sert à rien », répétant les préconisations de l’OMS qui finira par accepter le 30 avril 2021 ce que les scientifiques ont depuis longtemps démontré : le virus se transmet essentiellement par aérosol via l’air expiré et sur de longues distances5. En attendant d’autres solutions, le masque, s’il avait été disponible, aurait été bien utile !
Les variants
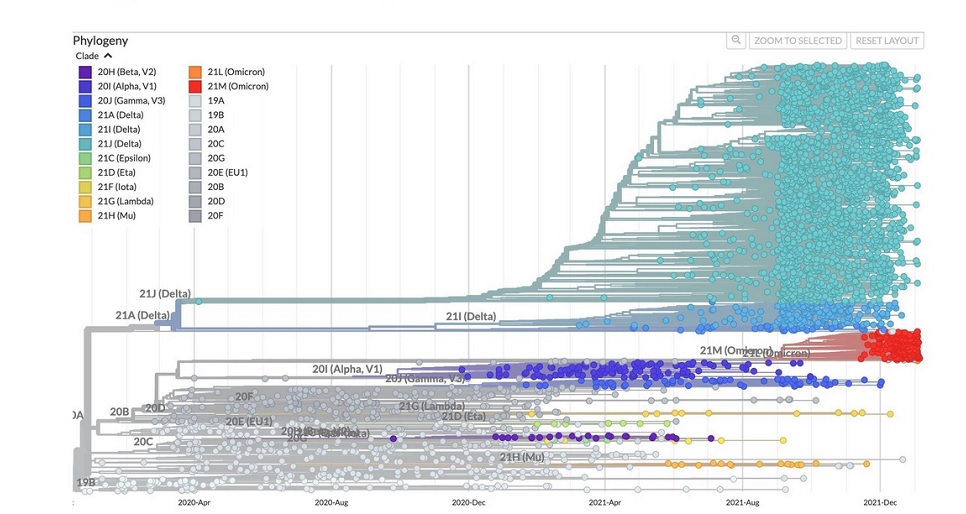
Depuis la souche originelle identifiée à Wuhan en novembre 2019, les variants se sont succédés : Alpha (B.1.17, Royaume-Uni, novembre 2020), Beta (B.1.351, Afrique du Sud, décembre 2020), Gamma (P.1, Brésil, décembre 2020), et Delta (B.1.617.2, Inde, novembre 2020). Cependant, au moment où le variant Delta pousse de nombreux non-vaccinés relativement peu âgés et des personnes âgées ou malades dans les hôpitaux, une nouvelle vague encore plus rapide déferle au début de l’année 2022 avec le variant Omicron (B.1.1.529) dont certaines souches sont parfois plus contagieuses que la varicelle et la rougeole, avec un R0 (nombre moyen de nouveaux cas qu’une seule personne contaminée va infecter) compris entre 5 et 24, soit près de 10 en moyenne6, du jamais vu chez ce type de virus ce qui, compte-tenu du délai très court entre la contamination et le début de la période contagieuse, en fait le virus le plus contagieux jamais identifié (voir encadré Omicron). En Afrique du Sud où il est apparu en novembre 2021, la vague n’aura duré que deux mois. En Europe, elle s’installe rapidement mais s’avérera moins létale que les précédentes. Omicron a cependant perduré jusqu’à aujourd’hui avec ses sous-variants et a donc infecté plus de monde et probablement causé plus de morts que tous les autres variants. Beaucoup plus contagieux que les variants précédents, Omicron est aussi plus apte à échapper à la protection immunitaire fournie par les vaccins ou l’immunité naturellement acquise. Avec son R0 remarquable, il a en quelque sorte joué le rôle d’un vaccin universel, ou du moins a permis à la population mondiale d’acquérir un seuil d’immunité collective suffisant pour que la maladie ne constitue plus une menace de santé publique, notamment en termes d’engorgement des hôpitaux comme ce fut le cas avant 2022. La douzaine de variants actuels dérivent presque tous de la souche JN.1 (septembre 2023), elle-même issue de l’Omicron BA.2.
Figure 1. Evolution des variants de SARS-CoV-2 jusqu’à Omicron en 2022. CC-BY-4.0 Emma Hodcroft – nextstrain.org
Omicron le variant à la surprenante évolution
Apparu en Afrique du Sud en novembre 2021, le variant Omicron continue de questionner. Sa contagiosité exceptionnelle reste inexplicable pour un coronavirus. Par rapport à la souche d’origine, il possède plus de 50 mutations dont 5 constituent une signature d’une adaptation à un hôte murin, alors qu’à l’origine SARS-CoV-2 n’était pas capable de se fixer aux récepteurs ACE2 du rongeur7. Cette donnée suggère qu’Omicron a évolué dans un environnement de type rongeur, que ce soit des cellules en culture ou dans la souris elle-même, naturellement ou dans un laboratoire8,9. Mais comment les souris ont-elles été infectées ? Comment le variant Omicron s’est-il ensuite adapté aux cellules humaines restent des questions sans réponse. Toutefois, une majeure partie de la communauté scientifique considère qu’Omicron aurait principalement évolué chez des patients humains immunodéprimés au cours d’une infection chronique qui aurait duré plusieurs mois10,11. Cette hypothèse pourrait également expliquer l’évolution des autres variants, puisque le variant Delta ne provient pas du variant Alpha, et Omicron ne dérive pas de Delta (voir Figure 1). Aucun ne suit une évolution classique dite « en échelle », telle celle qu’on observe par exemple chez le virus de la grippe H3N2 ou du rhume CoV-229, qui leur permet d’échapper à l’immunité. Pour Omicron, le doute subsiste quant à l’origine des mutations indiquant une adaptation à un hôte de type rongeur et son R0 hors norme questionne.
En France, les données précises sur la circulation du SARS-CoV-2 ne sont plus disponibles depuis la fermeture en juin 2023 de la plateforme SI-Dep (pour systèmes d’information de dépistage). Depuis plusieurs mois, les modèles de prévision ont été mis en pause alors que ceux concernant la grippe ont été longtemps actifs, attestant de la sévérité de l’épidémie grippale. Du point de vue physiopathologique, il existe une différence de taille entre l’avant et l’après Omicron : les premiers variants ciblaient préférentiellement les voies respiratoires basses (trachée, bronches, poumons), entraînant des infections graves. Omicron s’attaque plus spécifiquement aux voies respiratoires supérieures (du nez au larynx), ce qui le rend moins dangereux mais plus transmissible. Le port du masque dans les lieux publics reste rare mais non nul (13% des personnes ayant répondu à l’enquête CoViPrev en septembre 2024 déclarent le porter systématiquement). Les masques, surtout les respiratoires N95 ou FFP2, se sont avérés utiles pendant la deuxième vague12.
Actuellement, en dehors des immunodéprimés, la majorité des personnes infectées par le SARS-CoV-2 se soignent chez elles, comme pour une infection respiratoire virale classique. A l’hôpital, les traitements se sont banalisés et les malades, essentiellement des personnes âgées avec une pathologie sous-jacente, sont traités avec des anti-viraux, tel que le paxlovid. Les recherches sur les conséquences du Covid long, qui frappe au moins 2 millions de français (dont 10 à 15% ont des formes très sévères), s’intensifient. On sait que le virus persiste dans les organes qu’il a infectés (cerveau, système cardio-vasculaire, système immunitaire). Maintenant que cette maladie, dont les causes sont probablement multiples, est reconnue, il reste à prendre en charge le mieux possible les patients, parfois très lourdement atteints, et trouver des traitements et des alternatives leur offrant plus de confort, en attendant une hypothétique guérison13.
L’ONU avait alerté en mars 2020 sur les ravages qu’allait causer le SARS-CoV-2 dans les pays en développement. Fort heureusement, la catastrophe annoncée n’a pas eu lieu, même si, du fait du confinement, certains pays ont été durement touchés. L’Afrique est un bon exemple : sur plus d’un milliard d’habitants, elle a enregistré environ 1,5 million de cas de Covid-19 (selon les données de l’université John Hopkins), des chiffres bien inférieurs à ceux de l’Europe ou de l’Asie, qui pourraient s’expliquer par la jeunesse de sa population (âge médian de 19 ans) et le faible taux de personnes âgées (3% de plus de 65 ans), celles qui ont été les plus touchées dans les pays « riches », un climat et un mode de vie favorables à la non propagation du virus et des systèmes de santé communautaires éprouvés. Toutefois il ne s’agit que des cas confirmés, la plupart n’ayant pas été comptabilisés, ce qui donnerait une estimation avoisinant la centaine de millions.
La vaccinationet les effets indésirables
Rapidement, la solution du tout vaccin a été présentée comme étant nécessaire pour limiter la circulation du virus, le risque de développer des formes graves et engorger les hôpitaux. Elle est devenue quasiment obligatoire dans la plupart des pays occidentaux. En France, c’est le vaccin à ARN messager (ARNm) qui a été majoritairement injecté dès l’autorisation de mise sur le marché, fin 2020. Les deux premiers vaccins de ce type disponibles (Moderna et Pfizer/BioNtech) reposaient tous deux sur l’injection d’un ARN codant la protéine S (pour Spike), la protéine du virus lui permettant de se fixer sur les récepteurs présents à la surface des cellules de l’hôte infecté (récepteurs ACE2). D’autres vaccins, fabriqués de manière traditionnelle, à base de virus à ADN (tels Astra-Zeneka) ou de virus inactivé (Coronavac) ont aussi été utilisés. En France, la campagne de vaccination 2024/2025 utilise le vaccin monovalent à ARN messager Comirnaty JN1 de Pfizer/BioNTech, correspondant au variant JN.1 qui circule depuis septembre 2023.
La vaccination généralisée a indéniablement permis de sauver un grand nombre de vies, comme le montre un taux de létalité beaucoup plus faible chez les personnes vaccinées que chez les personnes non vaccinées. Cependant, la pertinence de sa généralisation aux enfants et aux adultes jeunes en bonne santé a été discutée14. En effet, dès février 2020, les premières publications chinoises avaient montré que les formes graves concernaient à plus de 80% les personnes âgés (65 ans et plus) et les malades avec des facteurs de comorbidités. De plus, au cours de la pandémie, seuls quelques enfants dans le monde seraient décédés directement du Covid 19. L’adhésion à la vaccination a largement fluctué depuis 2020 et une défiance persiste en France : en février 2024, 30% seulement des personnes âgées de 65 ans et plus (et seulement 10% des professionnels de santé) se sont fait inoculer le vaccin, alors que plus de la moitié (54%) se sont fait vacciner contre la grippe. Et pourtant, le SARS-CoV-2 a été plus mortel que la grippe (40 000 morts estimés pour le Covid 19 en 2022 contre 9 000 à 10 000 morts par an en moyenne pour la grippe). Depuis la mi-octobre 2024, la double vaccination grippe/Covid-19 est préconisée, bien que contrairement à la grippe dont le pic est hivernal, la circulation du SARS-CoV-2 s’effectue toute l’année. Une partie du public plus spécifiquement ciblé pense qu’elle est inutile. Cette défiance est multifactorielle. Citons quelques raisons :
La nouveauté de ce type de vaccin à ARN créée un doute légitime puisqu’il n’y a par définition comme pour toute innovation aucun recul. L’ARN peut-il gagner le noyau cellulaire, être « rétro-transcrit » (c’est-à-dire transformé en ADN) et s’insérer dans l’ADN, créant ainsi de possibles mutations ? Rappelons que près de 10% de notre patrimoine génétique correspond à des séquences virales acquises sur des millions d’années par transferts horizontaux de virus (à ADN ou ARN). De nombreux scientifiques pensent que cette hypothèse calquée sur l’action de rétrovirus (tel le VIH) ne s’applique pas à l’ARN du vaccin car il est fragile et ne devrait pas persister longtemps dans la cellule. Toutefois même si aucune donnée ne montre que l’intégration de l’acide nucléique du vaccin ARNm dans le noyau des cellules ait eu lieu, cela reste possible.
Des personnes vaccinées ont été contaminées et hospitalisées. Outre le fait qu’un vaccin n’est jamais efficace à 100%, l’échappement immunitaire est une des caractéristiques des variants issus d’Omicron, c’est-à-dire que certaines mutations de leur protéine Spike empêchent les anticorps de les reconnaitre et donc les neutraliser. De fait, l’efficacité vaccinale a fortement chuté. Au début 2022, seules les personnes âgées et vulnérables ainsi que les professionnels de santé étaient visées par la deuxième dose de rappel.
Le pass sanitaire entré en vigueur le 9 juin 2021, obligatoire dès 12 ans, et son caractère coercitif ont eu un impact très défavorable sur l’opinion du grand public.
Des scientifiques ont eux-mêmes suscité le doute en décrivant que la protéine S codée par le vaccin à ARN pouvait pénétrer dans le noyau de la cellule et inhiber les mécanismes de réparation de l’ADN (voir par exemple15, mais pour une raison inconnue l’article a été rétracté en mai 2022). Plusieurs généticiens, dont Axel Kahn, ont ainsi exhorté les pouvoirs publics à « éviter la vaccination des enfants car ce serait criminel ». En effet, la multiplication cellulaire chez les jeunes en pleine croissance est sans commune mesure avec celles des adultes et la non réparation potentielle d’erreurs de copie de l’ADN pourrait avoir des répercutions majeures sur leur développement. Mais au-delà de ces précautions, la communauté scientifique a reconnu qu’il n’y avait aucun bénéfice à vacciner et faire des rappels à des populations à faible risque comme les enfants.
Enfin, de nombreux effets secondaires de différents vaccins anti-SARS-CoV-2 ont été rapportés. Le dernier rapport publié par l’ANSM en août 2023 mentionnait 193 934 déclarations d’effets indésirables dont 25% de cas graves, pour plus de 156 788 000 injections (soit un peu plus de 1 personne/1000)16. Parmi les 25 potentiels évènements indésirables décrits, notons par ordre décroissant de fréquence : des troubles cardiovasculaires (péricardites, myocardites, AVC, infarctus du myocarde), des troubles neurologiques, des thromboses, des troubles articulaires, des troubles menstruels…). Dans le cas des myocardites, c’est une réponse généralisée de l’organisme au vaccin provoquant une inflammation du muscle cardiaque qui a été mise en cause et non la production d’anticorps. Des procédures pénales, très peu nombreuses, ont été engagées auprès de tribunaux, des indemnisations ont été accordées17.
Origine du SARS-CoV-2
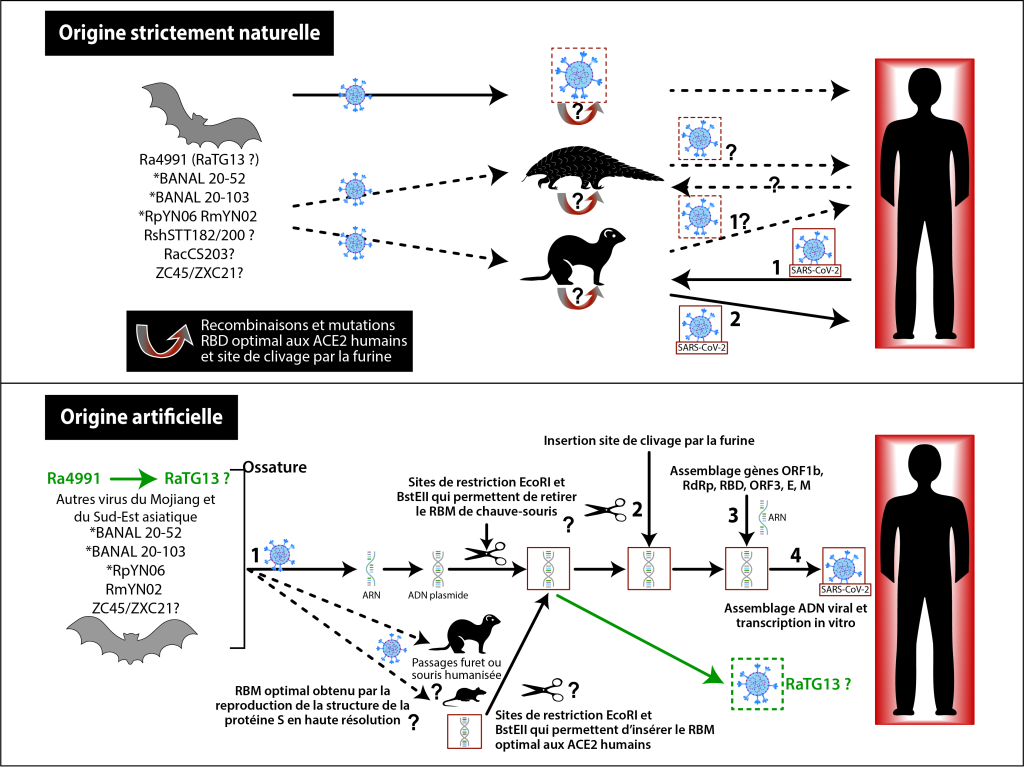
Au début de l’épidémie, l’origine zoonotique du SARS-CoV-2 était l’hypothèse privilégiée des experts du domaine. En effet, de nombreux virus de chauve-souris du Sud-Est asiatique se recombinent naturellement. SARS-CoV-2 aurait ainsi pu émerger au hasard de recombinaisons successives entre différents coronavirus de chiroptères avant d’être transmis directement à l’homme, ou via un animal hôte intermédiaire, à Wuhan où se situe le marché de Huanan qui héberge de nombreux animaux sauvages. Mais cette ville considérée comme le point de départ de l’épidémie est localisée à près de 2000 km des régions où vivent les chauves-souris qui hébergent des virus ressemblant à SARS-CoV-2. Force est de constater que 5 ans après le début de la pandémie, nous n’avons toujours pas trouvé le virus progéniteur de SARS-CoV-2, c’est à dire celui qui était présent dans un animal, juste avant d’être transmis à l’humain, et dont les séquences génétiques devraient être presque identiques (99,9%) à celles du SARS-CoV-2, comme cela a pu être observé chez les virus Zika ou Hendra ou SARS-CoV et MERS-CoV18. Nous ne savons pas plus où a précisément démarré l’épidémie à Wuhan, ni quel est l’animal responsable des premières contaminations humaines. Voyons ensemble les différentes pièces du puzzle et si l’hypothèse d’une origine liée aux laboratoires de recherche de Wuhan travaillant sur les coronavirus ne mérite pas d’être sérieusement évaluée.
L’hypothèse zoonotique
A la recherche des virus cousins naturels de SARS-CoV-2
. RaGT13, le virus de la grotte de Mojiang
Jusqu’en 2021, avec 96,2% d’identité, RaTG13 est décrit commele cousin naturel le plus proche de SARS-CoV-2. Ce virus a été isolé en 2013 dans la grotte minière de Mojiang (dans le Yunnan au Sud-ouest de la Chine) par les chercheurs du laboratoire de Wuhan spécialisé dans les virus de chauve-souris (WIV), cette grotte ayant été échantillonnée suite à l’infection en 2012 de travailleurs manipulant du guano de chauve-souris Rhinolophus affinis (Ra)19. Cependant, outre le fait que cette grotte est fermée aux observateurs étrangers depuis le début de la pandémie, plusieurs données questionnent sur l’origine strictement naturelle de RaTG13. En effet, il s’avère incapable de se fixer sur les récepteurs ACE2 des chauves-souris du genre Rhinolophus, censées être ses hôtes naturels20,21 ! De plus, sa protéine S possède dans sa région N terminale un domaine particulier de liaison aux gangliosides (des lipides) qui lui confère une plus grande infectiosité car elle lui permet de se fixer sur d’autres récepteurs qu’ACE2 ; mais la forme tridimensionnelle plate de ce domaine n’est présente chez aucun coronavirus hormis SARS-CoV-220.
En outre, seuls RaTG13 et SARS-CoV2 possèdent dans leur séquence ARN (constitué comme tous les ARN des 4 bases azotées A, U, G et C) des particularités qui signeraient une évolution convergente : tous les deux présentent une déplétion en dinucléotides CpG et un taux élevé de substitutions C>U22,23. L’explication est la suivante : lorsqu’une cytosine (C) se trouve en amont d’une guanine (CpG), elle peut être méthylée (par fixation d’un groupe méthyle CH3), ce qui favorise sa conversion (par désamination) en uracile (U), entraînant ainsi un taux élevé de substitution C>U. Ces particularités pourraient témoigner de pressions de sélection différentes de celles que rencontrent ces virus dans la nature, chez leur hôte naturel la chauve-souris. Elles pourraient résulter d’un nouvel environnement accélérant leur adaptation en favorisant l’accumulation de mutations. Comme l’ont montré de nombreuses données épigénétiques, un milieu perturbé ou stressant induit une forte méthylation des cytosines. Un tel environnement stressant à l’origine d’une forte sélection naturelle pourrait correspondre au passage de SARS-CoV-2 chez un nouvel hôte animal (tel le vison, comme cela a été observé depuis le début de l’épidémie18). Pour RaTG13, l’explication est peut-être à rechercher au niveau de la région N terminale de sa protéine S, qui ne lui permet pas de reconnaître les récepteurs de son hôte naturel de type chauve-souris, ce qui pourrait indiquer une adaptation à un hôte différent ou une évolution artificielle.
Enfin, il est important de signaler qu’une partie de RaTG13 (en particulier le gène RdRP) avait déjà été analysée en 2016, mais ce virus portait alors un autre nom (BtCoV/4991)24-25. Il s’agissait bien pourtant du même virus provenant de la mine du canton de Mojiang, à l’origine en 2012 d’une pneumonie atypique et foudroyante de trois travailleurs de la mine19. Cependant, il existe 5 différences entre la séquence publiée en 2016 et celle de 2020. Même si elles sont faibles (0,1%), elles sont difficiles à expliquer si le virus collecté en 2013 est resté au congélateur. Ainsi l’analyse poussée des séquences génétiques de RaTG13 interroge-t-elle toujours sur son origine.
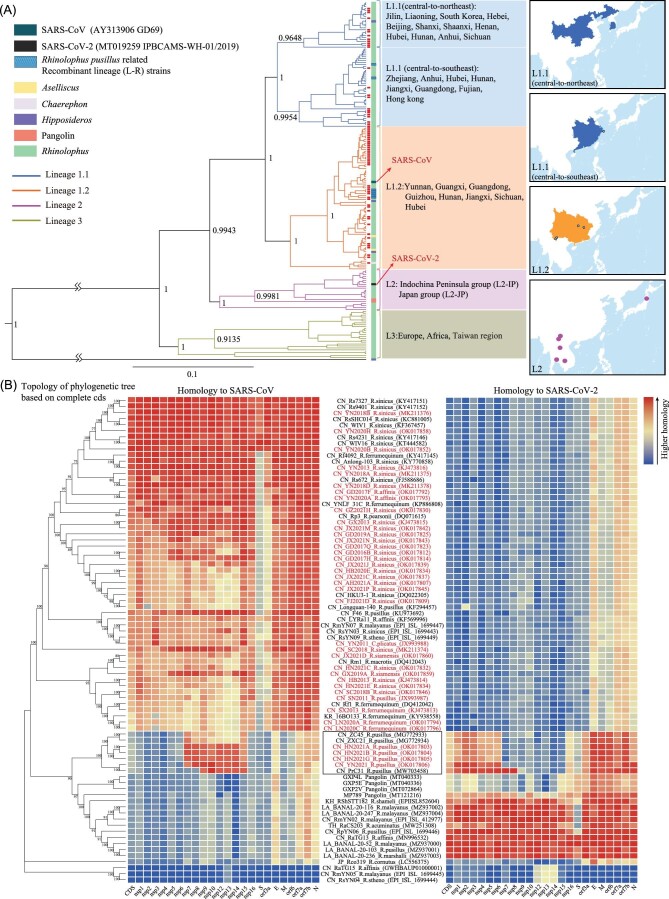
. RpYN06 et L-R, des virus du Sud de la Chine
En 2021, deux équipes publient leur découverte sur d’autres virus proches cousins de SARS-CoV-2 : d’une part, RpYN06, collecté également dans le Yunnan, sur la petite chauve-souris en fer à cheval, Rhinolophus pusillus. Avec ses 94,5% d’identité, RpYN06 devient le second cousin le plus proche de SARS-CoV-2 après RaTG13 27, avant qu’on ne découvre les virus BANAL du Laos (voir plus loin). D’autre part, suite à un travail mené sur plus de 36 000 chauve-souris de 100 espèces différentes, une nouvelle lignée de virus, appelée L-R, est décrite chez R. pusillus. Elle est le fruit d’une recombinaison naturelle entre la lignée L1, le groupe de virus apparentés à SARS-CoV, et la lignée L2, groupe de virus apparentés à SARS-CoV-228,29(voir Figures 2 et 3).
La petite chauve-souris en Fer à Cheval, Rhinolophus pusillus, espèce hôte de la lignée L-R. CC BY-NC 4.0 H.T.Cheng
Mais, malgré son envergure, cette étude n’a pas permis de trouver de plus proches cousins de SARS-CoV-2 ; elle montre cependant qu’en dehors de RaTG13, RmYN02 et RpYN06, les Sarbecovirus de la lignée 2 de SARS-CoV-2 sont rares en Chine.
. BANAL, des virus du Laos proches de SARS-CoV-2 mais sans site de clivage par la furine
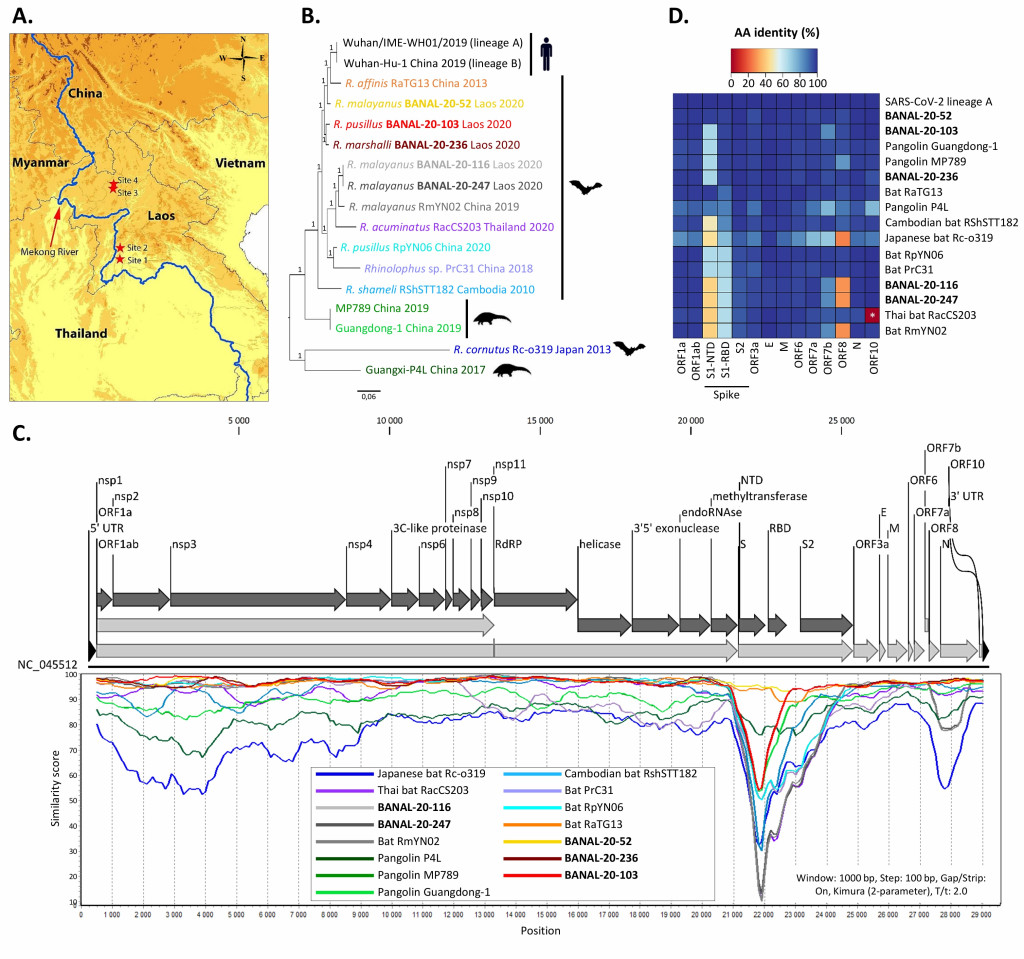
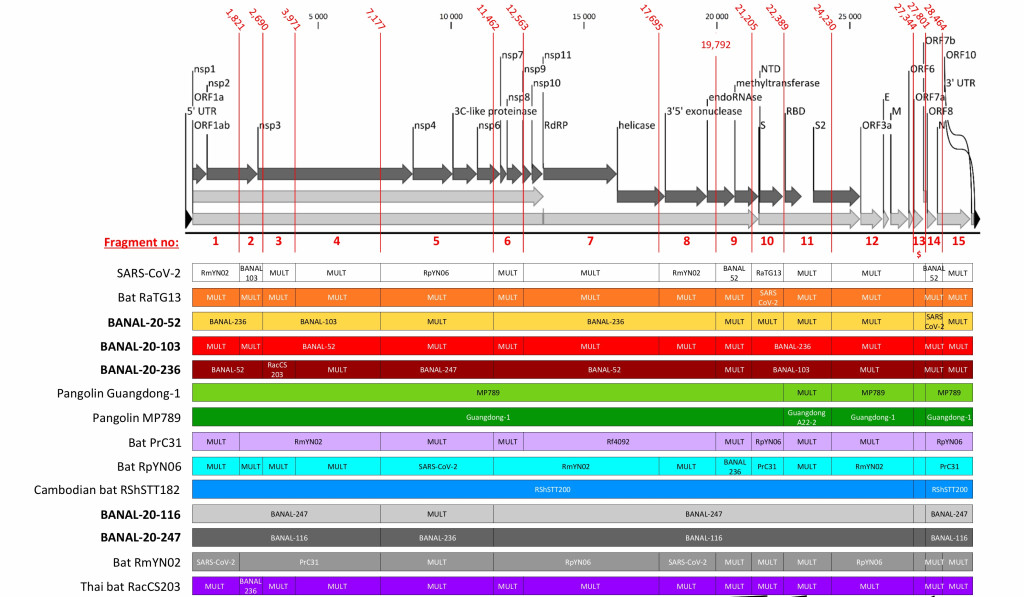
Nouveau rebondissement quand une équipe franco-laotienne (Instituts Pasteur de Paris et du Laos) décrit en février 2022 les virus BANAL (B pour bat, chauve-souris en anglais, et ANAL car isolés à partir de prélèvements rectaux)30,31. Les virus BANAL ne sont pas spécifiques d’une espèce de chauve-souris particulière puisque, par exemple, BANAL-52, BANAL-103 et BANAL-236 ont été trouvés respectivement chez R. malayanus, R. pusillus et R. marshalli. Avec près de 97% d’identité de séquences avec SARS-CoV-2, BANAL-52 s’avère être son plus proche cousin. De plus, les virus BANAL possèdent un domaine de liaison au récepteur humain ACE2 très proche de celui de SARS-CoV-2 (voir Figure 4). Cependant, comme le soulignent Virginie Courtier, généticienne de l’évolution à l’Institut Jacques Monod de Paris, et Etienne Decroly, virologue au CNRS d’Aix/Marseille, ces nouveaux virus sont dépourvus d’une séquence très caractéristique de SARS-CoV2, le site de clivage par la furine (voir encadré), restreignant leur capacité infectieuse (ce sont des virus entériques) et leur propagation inter-humaine32.
Figure 4. A gauche, a) lieu de collectes, b) relations de parentés des sarbecovirus de chiroptères, d’humains et de pangolins, c) taux d’homologie (en %) entre virus suivant la position des séquences génétiques, d) taux d’identité (en %) entre les acides aminés des 14 protéines (issues des gènes) de chaque virus. A droite, les fragments des virus issus de recombinaisons antérieures, avec indication pour chaque fragment du virus le plus proche ou de son origine multiple (« MULT »)30. Crédit : M. Eloit
Site de clivage par la furine
La protéine S du virus SARS-CoV-2 possède dans sa séquence en acides aminés un site particulier, appelé site de clivage par la furine : RRAR (R pour arginine et A pour alanine). Ce motif de 4 acides aminés n’est présent dans aucun des virus proches. Il confère au virus plusieurs avantages : une meilleure fixation au récepteur cellulaire ACE2, mais aussi à un autre récepteur cellulaire encore plus ubiquitaire, la neuropiline (NRP1 et NRP2) ; sa pénétration dans les cellules hôtes facilitée par des enzymes protéolytiques cellulaires (TMPRSS2 et TMPRSS13) 20. De nombreux virus possèdent des sites de clivage par la furine ; 132 ont été répertoriés. Cependant, chez l’animal, un seul virus naturel, l’alphacoronavirus félin, responsable de la péritonite infectieuse féline (FCoV-1), possède exactement la même séquence PRRAR que celle du SARS-CoV-233. Chez l’Homme, il existe également une protéine humaine (ENaC : Epithelial Na Channel) correspondant au canal sodium qui possède non seulement le même site furine (RRAR) que celui du SARS-CoV2 mais aussi les mêmes 4 acides aminés en aval (SVAS). Cette protéine située au niveau de la membrane des cellules épithéliales de différents organes (poumons, reins) peut entraîner une détresse respiratoire en cas de dysfonctionnement34.
Afin de savoir si ce site de clivage furine aurait pu émerger naturellement chez l’humain par le biais de mutations au fur et à mesure de sa propagation, des cellules humaines exprimant le récepteur ACE2 humain ont été infectées de manière sérielle par le virus BANAL-236. Après six cycles d’infection successifs – ce qui serait peu probable dans la nature car un virus à tropisme entérique cesserait de se propager rapidement – aucun site de clivage par la furine n’est apparu32,35. Ces données suggérent que ce site n’a pas pu émerger aisément dans la population humaine au cours d’une éventuelle phase précoce de circulation silencieuse du SARS-CoV-2, mais seulement au cours d’une recombinaison quasi contemporaine sur le marché de Wuhan au sein de l’espèce intermédiaire, une probabilité extrêmement faible voire quasi nulle. Son origine reste donc mystérieuse. La probabilité qu’il apparaisse spontanément a été estimée à 1 chance sur 321 milliards36, ce qui n’empêche pas qu’il puisse exister chez des virus de chiroptères qui n’ont pas encore été découverts ou qu’il ait été acquis après recombinaison entre deux virus lors d’une co-infection ou encore qu’il soit présent dans la séquence d’un virus d’un animal autre qu’une chauve-souris, tel l’alphacoronavirus félin avec lequel il se serait recombiné. Néanmoins, dans cette dernière hypothèse, il faudrait imaginer que la recombinaison n’ait concerné qu’une très courte séquence nucléotidique puisque seul le site de clivage par la furine et quelques nucléotides en amont et en aval du site sont présents dans la séquence du SARS-CoV-2.
. Virus vietnamiens et origine phylogéographique de SARS-CoV-2
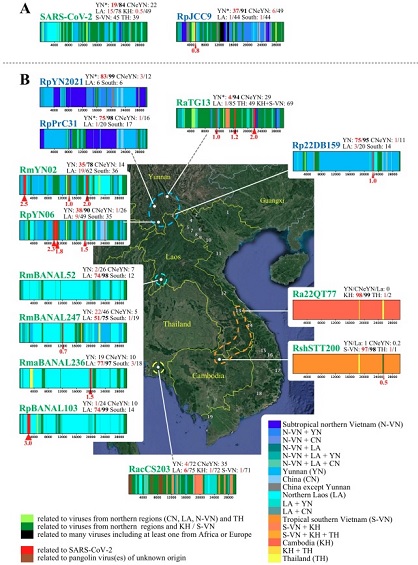
En 2023, Alexandre Hassanin, phylogénéticien du Muséum national d’histoire naturelle (MNHN), a entrepris une vaste étude phylogéographique pour élucider l’origine du SARS-CoV-2. Plus d’un millier de prélèvements de chauve-souris effectués au Vietnam entre 2017 et 2022 ont été analysés. Les résultats suggèrent que les différentes séquences génétiques composant SARS-CoV-2 proviendraient de virus de la région du triangle d’or, une région montagneuse d’Asie du Sud-Est aux confins du Laos, de la Birmanie et de la Thaïlande (qui pour certains inclut également une partie du Vietnam et le Yunnan chinois)37.De nombreux animaux sauvages y sont commercialisés sur les marchés traditionnels aux règles d’hygiène rudimentaires. Or le commerce d’espèces sauvages vivantes ainsi que les personnes fréquentant les grottes (chasse de chauves-souris, récolte de guano, tourisme…) favorisent la transmission zoonotique de coronavirus, ce qui est attesté notamment par la présence d’anticorps contre les β-coronavirus chez 1 à 4% de la population de la province du Yunnan avant l’épidémie de SARS-CoV-218. Cependant aucune épidémie de pneumonies mortelles n’y a jamais été répertoriée, même au cours de la pandémie de Covid-19 (les 3 mineurs décédés en 2012 dans la grotte de Mojiang n’ayant contaminé personne). Au Vietnam, deux nouveaux virus recombinants ont ainsi été mis en évidence, dont l’un possède un domaine de liaison aux récepteurs ACE2 humains très proche de celui du SARS-CoV-2 (voir Figure 5). Cette étude met aussi en lumière que les recombinaisons naturelles entre virus au Vietnam et au Laos semblent plus fréquentes dans la lignée de SARS-CoV-2 que ce qui a été décrit pour les virus chinois38.
Ainsi, cette publication confirme que des recombinaisons naturelles existent chez les virus SARs-CoV. Mais pour que le SARS-CoV-2 ait émergé naturellement, il aurait fallu au moins 7 événements de recombinaison entre des virus de chauve-souris30,31. Or, même dans la plus vaste étude jamais réalisée sur 100 espèces de chauve-souris en Chine, le seul clade, L-R, dans lequel des recombinaisons ont pu être observées, ne comporte que 2 événements majeurs de recombinaisons29.
Figure 5. Origines phylogéographiques des différents virus composant l’ossature de SARS-CoV-2 avec l’identification des séquences recombinées entre chaque virus 37. CC BY-SA Alexandre Hassanin
A la recherche du point de départ de l’épidémie de SARS-CoV-2
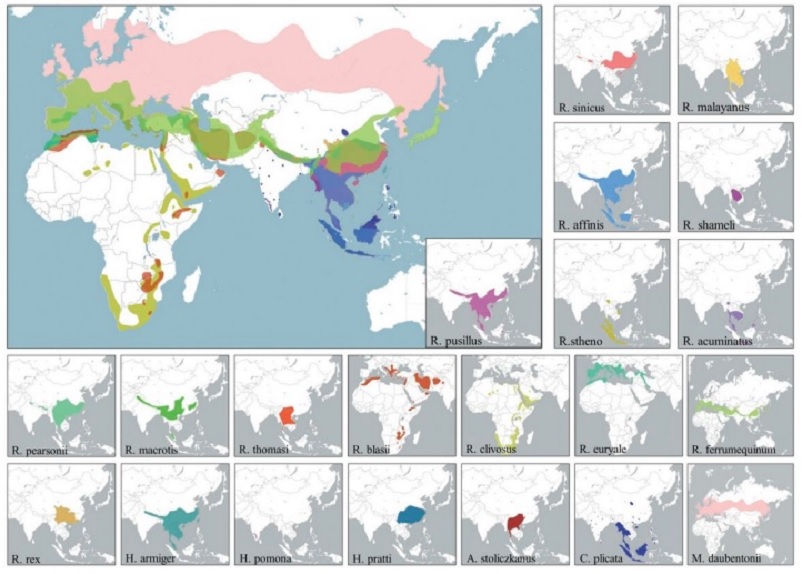
Les premiers virus SARS-CoV-2 identifiés officiellement en décembre 2019 provenaient du marché de Huanan à Wuhan, et semblaient appartenir à deux lignées (A et B) différant par deux mutations. La lignée B responsable des premières infections et la lignée A se répandant quelques semaines plus tard, sans qu’on sache si l’une dérivait de l’autre par mutation chez l’humain ou si chacune avait contaminé l’humain par l’intermédiaire d’hôtes différents39,40. Cependant, cette hypothèse reposait sur des modèles mathématiques qui se sont avérés erronés, obligeant les auteurs à publier un rectificatif, avant qu’une lettre de demande de rétractation de l’article finisse par remettre en question définitivement l’hypothèse de la double émergence41. Quoi qu’il en soit, comment SARS-CoV-2 (souche A ou B) a-t-il pu émerger naturellement à Wuhan alors que les virus BANAL les plus proches se trouvent à près de 2000 km et qu’ils sont les proches cousins mais en aucun cas des ancêtres directs de SARS-CoV-2 ? Et pourquoi aucun virus BANAL n’a-t-il jamais été décrit en Chine alors que leurs espèces hôtes de chauve-souris présentent une aire de répartition s’étendant jusqu’à 1000 km à l’intérieur du territoire chinois ? (Voir figure 6 avec les aires de répartition des chauve-souris abritant les virus retrouvés dans l’ossature génétique de SARS-CoV-2 (R. affinis, R. malayanus et R. pusillus).
Figure 6. Aires de répartitions des espèces de chauve-souris porteuses de Sarbecovirus28.
Outre l’origine de l’épidémie au marché de Huanan, de nombreux scientifiques remettent aussi en cause la date du début de l’épidémie41. En effet, plusieurs personnes semblent avoir été contaminées avant décembre 2019 sans avoir fréquenté le marché de Wuhan32. Par exemple, Connor Reed, un jeune gallois expatrié habitant Wuhan, présentait des symptômes de Covid 19 dès le 25 novembre 2019, soit deux semaines avant le premier cas communément admis correspondant à une vendeuse de crevettes du marché aux fruits de mer de Huanan42,43. De plus, de nombreux résultats suggèrent la présence du virus avant novembre 2019 en France, en Italie et au Brésil 44. Une contamination par des militaires américains porteurs du virus et qui l’auraient propagé à Wuhan lors des jeux militaires mondiaux d’octobre 2019 a également été évoquée par le régime chinois. Les témoignages de militaires français et étrangers rentrés malades de Wuhan en 2019 avant le début officiel de la pandémie restent inexpliqués18. Il est donc possible que les premières infections humaines aient eu lieu, entre le 23 octobre et le 8 décembre 2019 32, voire dès le mois d’août 2019.
Figure 7. 7ème jeux mondiaux militaires organisés à Wuhan en octobre 2019.
A la recherche du vecteur de propagation
Une autre question d’importance concerne le mécanisme par lequel SARS-CoV-2 s’est répandu. Le mystère reste entier. Plusieurs vecteurs animaux potentiels ont été évoqués : le pangolin malais (Manis javanica), la civette masquée (Paguma larvata) ou le chien viverrin (Nyctereutes procyonoides). Certes, de l’ADN de chien viverrin et l’ARN du SARS-CoV-2 ont été observés dans les prélèvements environnementaux effectués dans le marché de Huanan avant sa fermeture en janvier 202045. Mais tous les tests effectués sur les 457 animaux analysés issus de 18 espèces différentes se sont révélés négatifs, c’est-à-dire qu’ils ne contenaient pas de séquences du SARS-CoV-246. De plus, tous les ARNs de SARS-CoV-2 échantillonnés sur le marché sont identiques à ceux retrouvés chez des humains47. Or on sait qu’en passant d’une espèce hôte à une autre, le virus mute très vite. Si des animaux du marché avaient été porteurs du virus fin décembre 2019, il est donc plus probable qu’ils aient été contaminés par l’humain. De plus, il n’y pas eu d’épizootie observée chez ces espèces animales après l’émergence du virus en population humaine. Mais revenons au cas emblématique du pangolin.
Le 24 mars 2019, une vingtaine de pangolins malais, Manis javanica, sont recueillis vivants par un centre de la faune sauvage du Guangdong au Sud de la Chine, la plupart d’entre eux sont malades et meurent peu de temps après. L’autopsie révèle une infection pulmonaire. Les analyses des prélèvements seront publiées en octobre 2019 : ces pangolins, qui appartiennent à une espèce en danger critique d’extinction, donc extrêmement rare, seraient porteurs du virus Sendai issu de rongeurs et de différents autres virus dont des coronavirus48. Cependant il est important de rappeler que des virus qui donnent des formes aiguës et non chroniques ont très peu de chance de séjourner durablement chez des espèces strictement solitaires comme les pangolins malais. Pourtant, en février 2020, après le début de la pandémie, un nouveau coronavirus (pangolin CoV GD) est identifié49-51. Il présente une grande similarité de séquence avec celle de SARS-CoV-2 (97,5%) au niveau du domaine de liaison de la protéine Spike au récepteur (RBD), mais, sur l’ensemble de son génome, CoV GD ne partage que 88 à 93% avec SARS-CoV2. Un peu plus tard, une seconde souche de coronavirus spécifique du pangolin est découverte chez des spécimens provenant de la région de Guangxi (pangolin CoV GX). Sa séquence présente aussi une grande identité avec celle du SARS-CoV-2 au niveau du RBD52. Enfin, une troisième souche est identifiée (MP789) à partir d’autres pangolins malades interceptés par les services des douanes du Guandong. Sa séquence présente de potentielles recombinaisons au niveau du gène codant pour la protéine Spike avec les virus RaTG13 et, plus surprenant, avec ZC45/ZXC2153. Ce couple de virus de chauve-souris, isolé chez l’espèce Rhinolophus sinicus, dont l’aire de répartition est plus septentrionale que les autres espèces de rhinolophes impliquées jusqu’à présent, a été collecté dans la province du Zhejiang à l’Est de la Chine en 2015, à plus de 2000 km de toute présence de pangolins malais ! Son étude génétique, publiée d’abord par l’académie militaire chinoise en 201854, a été reprise en 2022 ; elle révèle des incohérences, suggérant que les séquences de ces virus auraient été mélangées à celles de virus de pangolin et/ou d’autres espèces animales55… La piste du pangolin a fait long feu…
L’hypothèse artificielle
L’hypothèse d’une origine zoonotique pouvait être considérée comme l’hypothèse la plus parcimonieuse au début de l’épidémie. Malheureusement, cinq ans après, ni le virus progéniteur de l’épidémie ni l’animal responsable des premières contaminations humaines n’ont été identifiés. Dans ce contexte, l’hypothèse d’une origine liée aux laboratoires de recherche travaillant sur les coronavirus présents dans la ville de Wuhan, en Chine, en collaboration avec les laboratoires américains de Caroline du Nord, doit être évaluée.
Le projet DEFUSE
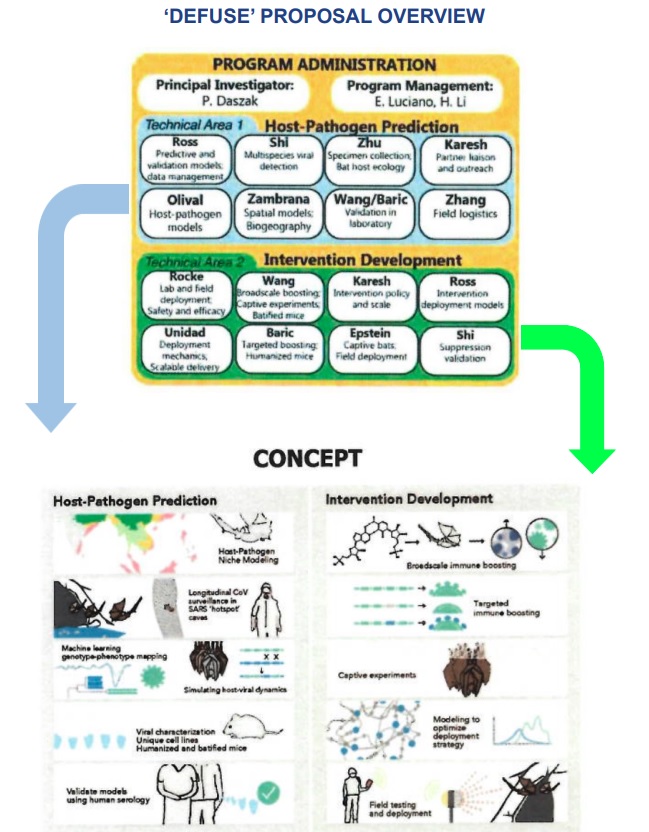
Dans le cadre de cette évaluation, il faut revenir sur les documents mis au jour en 2021 par le groupe Drastic (« équipe de recherche autonome radicale décentralisée »), un groupe informel d’enquêteurs autodidactes et de scientifiques travaillant dans les domaines de la microbiologie et de la génétique56,57. Ces documents concernent une demande de financement, le projet DEFUSE58,59, que l’ONG EcoHealthAlliance a effectuée en 2018 auprès de la DARPA (Agence du Département de la Défense du Pentagone aux USA). L’intention de ces auteurs, incluant Peter Daszak (zoologue, président de l’ONG), Ralph Baric (virologue, de l’université de Caroline du Nord), et la virologue Zengli Shi de l’Institut de virologie de Wuhan (WIV) en Chine, consistait à créer un coronavirus chimérique avec une ossature de virus apparentées au SARS-CoV en y insérant artificiellement deux séquences : 1) celles codant la protéine S issue des coronavirus de chauve-souris du sud-est asiatique pour lesquels le domaine de liaison au récepteur (RBD) s’adapte parfaitement aux récepteurs ACE2 des cellules humaines; 2) celles codant un site de clivage par la furine d’origine humaine qui facilite la fusion du virus avec les cellules infectées. De telles expériences, dites de « gain de fonction », ont clairement pour but, même si leurs auteurs s’en défendent60, de créer un virus dont la transmission et la virulence sont augmentées chez les humains par rapport aux virus apparentés trouvés dans la nature.
Figures 8. Documents mis au jour par le groupe Drastic sur le projet DEFUSE soumis au pentagone en 2018 impliquant la Chine et les Etats-Unis. Source publique
La demande de financement DEFUSE n’a pas été retenue par le DARPA et les révélations ne démontrent aucunement que les expériences proposées ont effectivement été réalisées. Cependant, il est important de préciser d’une part que ce projet proposait d’effectuer ces expériences de gain de fonction en partie à Wuhan en Chine et en Caroline du Nord aux Etats-Unis ; d’autre part, que les virus ainsi augmentés mais atténués, agissant comme des « vaccins », auraient pu être administrés directement in situ, via des aérosols, sur les colonies de chauve-souris « à risque » se reposant dans leurs grottes. Et si tout ceci peut ressembler à de la science-fiction, certaines de ces expériences ont été réalisées, notamment la création du fameux site de clivage par la furine si particulier au SARS-CoV-2.
Un site de clivage plutôt RRAR ?
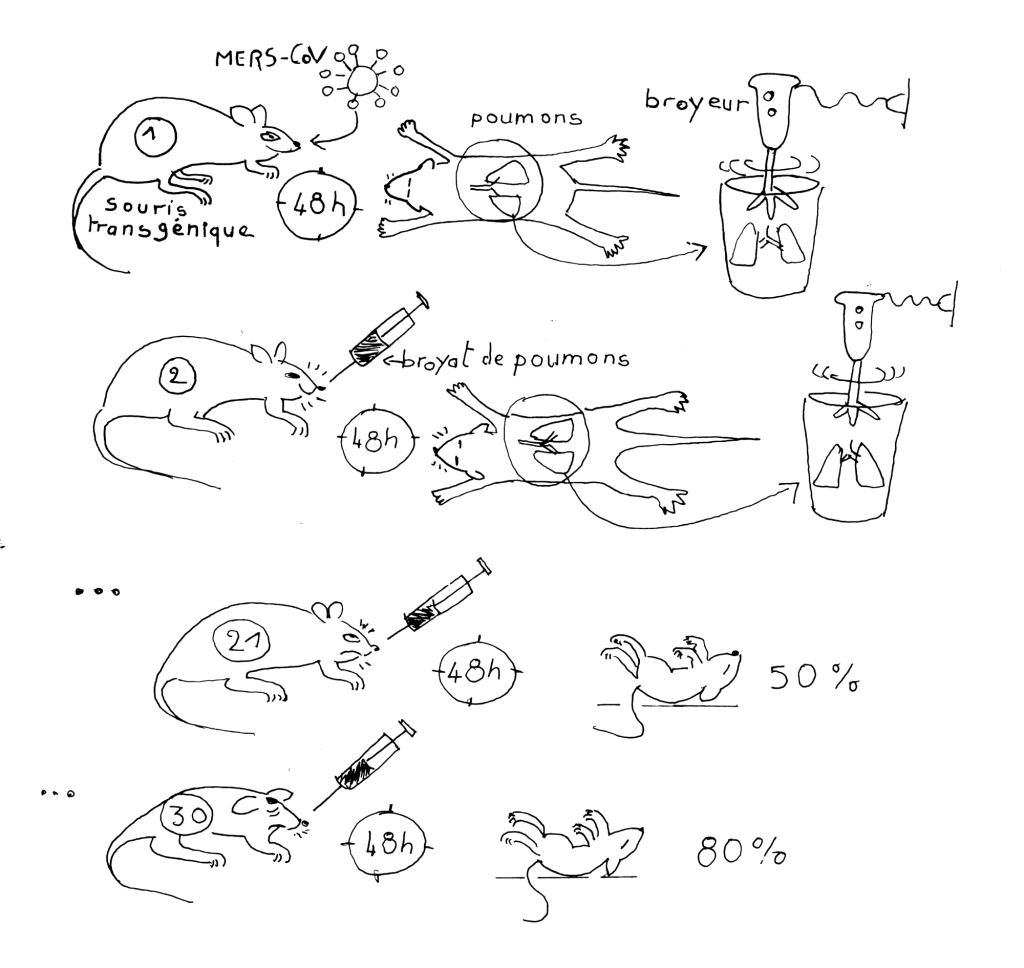
Très récemment, en décembre 2024, Lisewski61 a montré que le site furine du SARS-CoV-2 (PRRAR) que l’on retrouve naturellement dans l’alphacoronavirus de la péritonite infectieuse féline (FCoV-1)33 est également présent presque à l’identique (PRRVR) dans un MERS rendu artificiellement très virulent par passages successifs dans des souris transgéniques « humanisées ». (voir figure 9).
Figure 9. Expériences sur des souris transgéniques conçues et réalisées à l’université de l’Iowa (Etats-Unis) avant 2017 par le virologue chinois Kun Li, qui avait effectué sa thèse entre 2005 et 2010 au WIV62. Les souris transgéniques qu’il a employées expriment à la surface de leurs cellules un récepteur humain permettant la fixation du coronavirus hCoV-EMC, responsable d’une épidémie humaine fatale en 2012. Le virus est pulvérisé dans les narines de ces souris puis se réplique dans leurs poumons sans causer de maladie. Deux jours plus tard, elles sont sacrifiées, leurs poumons broyés et la préparation est de nouveau inoculée à des souris transgéniques. Les cycles sont répétés. Au fur et à mesure des passages, la préparation virale s’enrichit en virus porteurs de mutations facilitant leur fixation au récepteur ; au 30e passage, un clone viral est sélectionné, il est mortel pour les souris62. Au cours de l’expérience, la séquence d’acides aminées du site de clivage furine du virus est passée de PRSVR à PRRVR.
CC BY-NC-SA 4.0 Nicole Morello
Officiellement ce type d’expériences est justifié par leurs auteurs afin de disposer de modèles animaux utiles pour tester des vaccins contre de nouveaux virus. La découverte de Lisewski montre qu’un site de clivage par la furine aurait pu être créé et/ou inséré artificiellement au cours de manipulations génétiques. Mais ce site pourrait aussi provenir de la protéine humaine (ENaC : Epithelial Na Channel) qui possède exactement le même motif d’acides aminés que celui de SARS-CoV-2 (RRAR) avec les mêmes 4 acides aminés situés en aval. Cette protéine fait l’objet d’études menées depuis de nombreuses années par le laboratoire de l’université de Caroline du Nord de Ralph Baric à Chapel Hill aux Etats-Unis. Elle joue un rôle dans l’homéostasie du liquide de surface des voies respiratoires. Son dérèglement est associé aux affections respiratoires telles que la mucoviscidose, où le fonctionnement d’ENaC est altéré du fait de mutations génétiques. Le site de clivage furine de SARS-CoV-2 possède le même motif que l’ENaC et permet l’action protéolytique de la furine humaine63. Cette identité avec le site furine de la protéine ENaC questionne car elle correspond exactement aux intentions affichées dans le projet DEFUSE qui prévoyait d’insérer des sites de clivage trouvés chez l’homme « we will introduce appropriate human-specific cleavages sites and evaluate growth potential in Vero cell and HAE cultures. » (paragraphe « S2 Proteolytic Cleavage and Glycosylation Sites » page 11 du projet DEFUSE58.
Ossature du SARS-CoV2
Les expériences de gain de fonction proposées dans le projet DEFUSE déposé en 2018 ne pouvaient concerner que les virus collectés ou publiés avant 2018 (lignée SARS-CoV-1). La seule certitude que nous ayons à l’heure actuelle est que parmi tous les virus très proches de SARS-CoV-2 (RaTG13, RmYN02, RPYN06, BANAL 52, BANAL 103), seul RaTG13, aux caractéristiques hors normes pour un virus naturel, avait déjà été collecté en 2013 et conservé au WIV. Mais s’il ne s’agit pas d’expériences volontaires de manipulations génétiques, il est aussi possible que les chercheurs de Wuhan aient été contaminés accidentellement par le virus SARS-CoV-2 ou son géniteur direct, lors de la collecte de chauve-souris dans la nature ou au cours de l’étude en laboratoire des échantillons collectés, ou qu’ils aient cultivé dans des conditions favorables des prélèvements de chauve-souris infectés par deux coronavirus différents qui se seraient recombinés, générant par hasard un site de clivage par la furine.
Crédit : Vera Makina, Le Canard enchainé, n°5421, 2 octobre 2024
Pour résumer, que SARS-CoV-2 soit issu de manipulations génétiques volontaires (construction d’un virus chimérique avec des séquences virales en partie non déclarées ou publiées), ou qu’il résulte d’expériences non programmées, il faut imaginer qu’un accident de laboratoire – ou une fuite délibérée – s’en soit suivi, permettant sa propagation dans la nature (Wuhan), propagation intra-humaine ou via une espèce animale intermédiaire. Une succession d’événements qui a paru suffisamment improbable pour que la piste naturelle (zoonose) soit, un temps, privilégiée. Pourtant des fuites accidentelles de virus de laboratoires se sont déjà produites, comme pour la grippe russe de 1977 ou le SARS en 2003, fuites qui peuvent d’autant mieux s’expliquer que les conditions de manipulation des virus sont inadaptées, ce qui est avéré au WIV25.
Conclusion
Depuis notre dernière publication en 2021 faisant un état des lieux sur l’origine naturelle ou artificielle du Covid 1918, de nombreuses études sont parues dont nous avons fait la synthèse dans le présent article. Mais, en dépit de leur abondance et de leur qualité, il n’existe actuellement aucune preuve définitive permettant de trancher entre ces hypothèses. En revanche, ce qui est sûr c’est qu’un climat délétère s’est installé, jetant le doute sur les activités scientifiques en général, et des laboratoires chinois ou américains impliqués dans les expériences de gain de fonction en particulier. Ces doutes témoignent de la perte de confiance du public vis à vis des médias ou des groupes de pression qui pour la plupart relaient sans le recul nécessaire et sans connaissance approfondie des informations scientifiquement fausses, telle la piste du pangolin ou du chien viverrin comme hôtes intermédiaires, et celle du marché de Huanan de Wuhan comme point de départ de l’épidémie. Une partie de la communauté scientifique elle-même n’a pas, un temps, souhaité contredire la théorie d’une origine naturelle du virus, comme l’illustre la lettre publiée dans The Lancet, dans laquelle 27 scientifiques soutenaient la Chine pour sa transparence (!) et concluaient : « Nous sommes unis pour condamner fermement les théories du complot suggérant que le Covid-19 n’a pas une origine naturelle« 64. Rappelons que Peter Daszak, directeur de l’ONG EcoHealth Alliance, membre de l’OMS, et coordonnateur de la lettre travaillait avec le WIV à une période (2014 et 2017) où le moratoire empêchait les Etats-Unis de réaliser des expériences de gain de fonctions18,42. En réponse à cette lettre, plusieurs scientifiques français et étrangers, se sont insurgés, réclamant une liberté de la démarche scientifique et s’opposant fermement à l’idée qu’ils étaient des amateurs ou des … complotistes ! 18,42,56, un jugement toujours d’actualité dans la presse57,65,66 : « La recherche de l’origine du Covid-19 mérite mieux que des théories du complot … qui alimentent en retour les attaques anti-science » 67.
Figure 10. Schéma des hypothèses naturelles et artificielles de l’origine de SARS-CoV-2 18.
Pour conclure, un virus a émergé selon toute vraisemblance à Wuhan en Chine entre les mois d’août et de novembre 2019. Son ossature génétique est une mosaïque de différents virus portés par des chauves-souris chinoises et du Sud-Est asiatique. Une de ses propriétés génétiques est inédite : la présence d’un site de clivage par la furine dont le motif ressemble à celui retrouvé dans la nature chez un coronavirus félin et à celui d’un MERS-CoV sélectionné artificiellement, et est identique à celui de la protéine humaine ENaC dont le rôle est corrélé à certaines détresses respiratoires et affections digestives. L’origine de ce virus et de son dernier variant, Omicron, qui a mis fin à la sévérité de la pandémie il y a trois ans, reste toujours mystérieuse : provient-il uniquement de la nature ou bien a-t-il été adapté à l’humain par des manipulations en laboratoire68 (voir figure 10), une hypothèse que soutiennent actuellement les scientifiques du FBI, le service fédéral du renseignement allemand69 et une partie du Secret Intelligence Service britannique ? En France, seule l’académie de médecine penche pour un accident de laboratoire70, avec plusieurs scientifiques, dont le virologue Simon Wain-Hobson71, qui par mesure de précautions préconisent un débat sur les expériences de gain de fonction sur les virus car elles font peser sur la population mondiale un danger sans commune mesure avec les bienfaits hypothétiques d’expériences destinées à contrer de futures maladies.
Remerciements à Virginie Courtier-Orgogozo, biologiste spécialisée en évolution et génétique, Directrice de Recherche CNRS et Professeure associée à l’Ecole Polytechnique, Institut Jacques Monod et Renaud Piarroux, médecin épidémiologiste, Professeur à Sorbonne Université et Chef de service à l’hôpital de la Pitié-Salpêtrièrepour leur relecture, mais qui ne partagent pas forcément tous les points de vue développés par les auteurs dans cet article.
Morello, D. et H. Cap. 2021. Covid-19, de l’insouciance aux interrogations. Un tour d’horizon sur les vaccins, les variants et l’origine du SARS-CoV-2. Rubrique Parlons sciences du muséum de Toulouse, Juin 2021.
Liu, Y et Rocklöv J, The effective reproductive number of the Omicron variant of SARS-CoV-2 is several times relative to Delta, Journal of Travel Medicine, Volume 29, Issue 3, April 2022, taac037, https://doi.org/10.1093/jtm/taac037
Piplani S, Singh PK, Winkler DA, Petrovsky N. 2021? In silico comparison of SARS-CoV-2 spike protein-ACE2 binding affinities across species and implications for virus origin. Sci Rep. 2021 Jun 24;11(1):13063.
Sun, Y, Lin W, Dong W, Xu, J. Origin and evolutionary analysis of the SARS-CoV-2 Omicron variant. J. Biosafety Biosecur., 4 (1) (2022), pp. 33-37
Wei C, Shan K-J, WangW, Zhang S, Huan Q, Qian W. Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. J Genet Genomics (2021) pp.1111-1121. doi: 10.1016/j.jgg.2021.12.003. Epub 2021 Dec 24.
Bolis, M., Uceda Renteria, S., Alagna, L. et al. SARS-CoV-2 genomic evolution during a severe and long-lasting omicron infection under antiviral therapy. BMC Infect Dis25, 359 (2025). https://doi.org/10.1186/s12879-025-10740-w
Rahalkar MC, Bahulikar RA. 2020. Lethal pneumonia cases in mojiang miners (2012) and the mineshaft could provide important clues to the origin of SARS-CoV-2. Front Public Health. (2020) 8:581569. 10.3389/fpubh.2020.581569
Segreto, R., Y. Deigin, K. Mccairn, A. Sousa, D. Sirotkin, K. Sirotkin, J.J. Couey, A. Jones & D. Zhang. 2021. – Should we discount the laboratory origin of COVID-19? Environmental Chemistry Lettershttps://doi.org/10.1007/s10311-021-01211-0
Tang, X., C. Wu1, X. Li, Y. Song, X. Yao, X. Wu, Y. Duan, H. Zhang, Y. Wang1, Z. Qian, J. Cui & J. Lu. 2020. – On the origin and continuing evolution of SARS-CoV-2. National Science Review, 7: 1012–1023.
Matyacek, R., & A. Kovarik. 2020. – Mutation Patterns of Human SARS-CoV-2 and Bat RaTG13 Coronavirus Genomes Are Strongly Biased Towards C>U Transitions, Indicating Rapid Evolution in Their Hosts. Genes, 11, 761-774.
Simmonds, P. 2020.– Rampant C>U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences COVID 19 for Their Short- and Long-Term Evolutionary Trajectories. mSphere 5:e00408- 20. https://doi.org/10.1128/mSphere.00408-20
Ge XY, Wang N, Zhang W, Hu B, Li B, Zhang YZ, Zhou JH, Luo CM, Yang XL, Wu LJ, Wang B, Zhang Y, Li ZX, Shi ZL. 2016. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol Sin. 2016 Feb;31(1):31-40. doi: 10.1007/s12250-016-3713-9. Epub 2016 Feb 18. PMID: 26920708; PMCID: PMC7090819.
Segreto, R. & Y. Deigin. 2020. – The genetic structure of SARS-CoV-2 does not rule out a laboratory origin. BioEssays 202000240, 1-9.
Zhou H, Ji J, Chen X, Bi Y, Li J, Wang Q, Hu T, Song H, Zhao R, Chen Y, Cui M, Zhang Y, Hughes AC, Holmes EC, Shi W. 2021. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell. 2021 Aug 19;184(17):4380-4391.e14. doi: 10.1016/j.cell.2021.06.008.
Zhou H, Ji J, Chen X, Bi Y, Li J, Wang Q, Hu T, Song H, Zhao R, Chen Y, Cui M, Zhang Y, Hughes AC, Holmes EC, Shi W. 2021. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell. 2021 Aug 19;184(17):4380-4391.e14. doi: 10.1016/j.cell.2021.06.008.
Wu Z, Han Y, Wang Y, et al. 2021. A comprehensive survey of bat sarbecoviruses across China for the origin tracing of SARS-CoV and SARS-CoV-2. Research Square; 2021. DOI: 10.21203/rs.3.rs-885194/v1.
Wu Z, Han Y, Wang Y, Liu B, Zhao L, Zhang J, Su H, Zhao W, Liu L, Bai S, Dong J, Sun L, Zhu Y, Zhou S, Song Y, Sui H, Yang J, Wang J, Zhang S, Qian Z, Jin Q. 2022. A comprehensive survey of bat sarbecoviruses across China in relation to the origins of SARS-CoV and SARS-CoV-2. Natl Sci Rev. 2022 Oct 11;10(6):nwac213. doi: 10.1093/nsr/nwac213. PMID: 37425654; PMCID: PMC10325003.
Temmam, S., Vongphayloth, K., Baquero, et al. 2021. Coronaviruses with a SARS-CoV-2-like receptor[1]binding domain allowing ACE2-mediated entry into human cells isolated from bats of Indochinese peninsula. Research square September 17th, 2021. DOI: https://doi.org/10.21203/rs.3.rs-871965/v1
Temmam, S., Vongphayloth, K., Baquero, E. et al. 2022. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 604, 330–336 (2022). https://doi.org/10.1038/s41586-022-04532-4
Courtier V et Decroly E. 2022. Origine du SARS-CoV–2 et duCovid-19: le point sur l’enquête en cours et les dernières hypothèses. The conversation septembre 2022
Praveen Anand, Arjun Puranik, Murali Aravamudan, AJ Venkatakrishnan, Venky Soundararajan (2020) SARS-CoV-2 strategically mimics proteolytic activation of human ENaC eLife 9:e58603
Temmam S, Montagutelli X, Herate C, Donati F, Regnault B, Attia M, Baquero Salazar E, Chretien D, Conquet L, Jouvion G, Pipoli Da Fonseca J, Cokelaer T, Amara F, Relouzat F, Naninck T, Lemaitre J, Derreudre-Bosquet N, Pascal Q, Bonomi M, Bigot T, Munier S, Rey FA, Le Grand R, van der Werf S, Eloit M. 2023. SARS-CoV-2-related bat virus behavior in human-relevant models sheds light on the origin of COVID-19. EMBO Rep. 2023 Apr 5;24(4):e56055. doi: 10.15252/embr.202256055. Epub 2023 Mar 6. PMID: 36876574; PMCID: PMC10074129.
Ambati BK, Varshney A, Lundstrom K, Palú G, Uhal BD, Uversky VN and Brufsky AM. 2022 MSH3 Homology and Potential Recombination Link to SARS-CoV-2 Furin Cleavage Site. Front. Virol. 2:834808. doi: 10.3389/fviro.2022.834808
Hassanin, A., Tú, V. T., Görföl, T., LAM, N. Q., Pham, P., Chu, H., … & Wurtzer, S. 2023. Phylogeographic evolution of horseshoe bat sarbecoviruses in vietnam and implications for the origins of sars-cov and sars-cov-2. https://doi.org/10.21203/rs.3.rs-3227228/v1
Hassanin, A., Tu, V. T., Görföl, T., Ngon, L. Q., Pham, P. V., Hang, C. T., Tuan, T. A., Prot, M., SimonLorière, E., Kemenesi, G., Tóth, G. E., Moulin, L., & Wurtzer, S. 2024. Phylogeography of horseshoe bat sarbecoviruses in Vietnam and neighbouring countries. Implications for the origins of SARS-CoV and SARS-CoV-2. Molecular Ecology, 33, e17486. https://doi.org/10.1111/mec.17486
Pekar, Jonathan E., Andrew Magee, Edyth Parker, Niema Moshiri, Katherine Izhikevich, Jennifer L. Havens, Karthik Gangavarapu, et al. 2022. “The Molecular Epidemiology of Multiple Zoonotic Origins of SARS-CoV-2.” Science 377 (6609): 960–966.
Worobey, Michael, Joshua I. Levy, Lorena Malpica Serrano, Alexander Crits-Christoph, Jonathan E. Pekar, Stephen A. Goldstein, Angela L. Rasmussen, et al. 2022. “The Huanan Seafood Wholesale Market in Wuhan Was the Early Epicenter of the COVID-19 Pandemic.” Science 377 (6609): 951–959.
Perrier B. 2021. SARS-CoV-2 Aux origines du Mal. Belin
Audureau W. 2024. Le fragile témoignage de Connor Reed, l’expatrié qui aurait attrapé le Covid-19 avant tout le monde. Le monde 26/11/2024
Canuti M, Bianchi S, Kolbl O, et al. 2022. Waiting for the truth: is reluctance in accepting an early origin hypothesis for SARS-CoV-2 delaying our understanding of viral emergence? BMJ Glob Health 7.
Crits-Christoph A, Levy JI, Pekar JE, Goldstein SA, Singh R, Hensel Z, Gangavarapu K, Rogers MB, Moshiri N, Garry RF, Holmes EC, Koopmans MPG, Lemey P, Peacock TP, Popescu S, Rambaut A, Robertson DL, Suchard MA, Wertheim JO, Rasmussen AL, Andersen KG, Worobey M, Débarre F. 2024. Genetic tracing of market wildlife and viruses at the epicenter of the COVID-19 pandemic. Cell. 2024 Sep 19;187(19):5468-5482.e11. doi: 10.1016/j.cell.2024.08.010.
Gao, G, Liu W, Liu P, et al. 2023. Surveillance of SARS-CoV-2 in the environment and animal samples of the Huanan Seafood Market. In Review 2022. doi:10.21203/rs.3.rs-1370392/v1
Bloom, J. 2023. Association between SARS-CoV-2 and metagenomic content of samples from the Huanan Seafood Market. bioRxiv preprint doi: https://doi.org/10.1101/2023.04.25.538336
Liu P, Chen W, Chen JP. Viral metagenomics revealed sendai virus and coronavirus infection of malayan pangolins (Manis javanica). Viruses. 2019;11(11). doi:10.3390/v11110979
Wong MC, Cregeen SJJ, Ajami NJ, Petrosino JF. Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019. bioRxiv. 2020;2013:2020.02.07.939207. doi:https://doi.org/10.1101/2020.02.07.939207
Zhang T, Wu Q, Zhang Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr Biol. 2020;30(7):1346-1351.e2. doi:10.1016/j.cub.2020.03.022
Xiao K, Zhai J, Feng Y, et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. 2020;583(7815):286-289. doi:10.1038/s41586-020-2313-x
Lam TTY, Jia N, Zhang YW, et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020;583(7815):282-285. doi:10.1038/s41586-020-2169-0
Liu P, Jiang J-Z, Wan X-F, Hua Y, Li L., Zhou J, Wang, X, Hou, F, Chen, J, Zou, J, Chen, J. 2020. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLOS Pathogens, 16 (5), e1008421. https://doi.org/10.1371/journal.ppat.1008421
Hu, D., C. Zhu, L. Ai, T. He, Y. Wang, F. Ye, L. Yang, C. Ding, X. Zhu, R. Lv, J. Zhu, B. Hassan, Y. Feng, W. TAn & C. Wang. 2018. – Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerging Microbes Infections, 7: 154.
Jones, A.; Massey, S.E.; Zhang, D.; Deigin, Y.; Quay, S.C. 2022. Forensic Analysis of Novel SARS2r-CoV Identified in Game Animal Datasets in China Shows Evolutionary Relationship to Pangolin GX CoV Clade and Apparent Genetic Experimentation. Appl. Microbiol. 2022, 2, 882–904. https://doi.org/10.3390/ applmicrobiol204006
Coq-Chodorge C. et Massey J. 2021. Hypothèse d’une fuite de labo : les États-Unis au cœur de l’enquête sur l’origine du Covid-19. Médiapart 15 juillet 2021.
Audureau W. 2025. Covid-19 : comment un groupe d’enquêteurs amateurs a poussé la thèse d’un virus sorti d’un laboratoire. Le monde 17/02/2025
Li K, Wohlford-Lenane CL, Channappanavar R, Park JE, Earnest JT, Bair TB et al. Mouse-adapted MERS coronavirus causes lethal lung disease in human DPP4 knockin mice. 2017. Proc Natl Acad Sci U S A 114E3119
Praveen Anand, Arjun Puranik, Murali Aravamudan, AJ Venkatakrishnan, Venky Soundararajan (2020) SARS-CoV-2 strategically mimics proteolytic activation of human ENaC eLife 9:e58603
Calisher, C., D. Carroll, R. Colwell, R. B. Corley, P. Daszak, C. Drosten, L. Enjuanes, J. Farrar, H. Field, J. Golding, A. Gorbalenya, B. Haagmans, J. M. Hughes, W. B. Karesh, G. T. Keusch, S.K. Lam, J. Lubroth, J.S. Mackenzie, L. Madoff, J. Mazet, P. Palese, S. Perlman, L. Poon, B. Roizman, L. Saif, K. Subbarao & M. Turner. 2020. – Statement in support of the scientists, public health professionals, and medical professionals of China combatting COVID-19. The Lancet, 395 : 42-43.
Perrier B. 2023. L’obscurantisme au pouvoir. Editions Max Milot
Debarre F. 2025. La pandémie de Covid-19pourrait-elle avoir commencé dans un laboratoire ? The conversation janvier 2025.
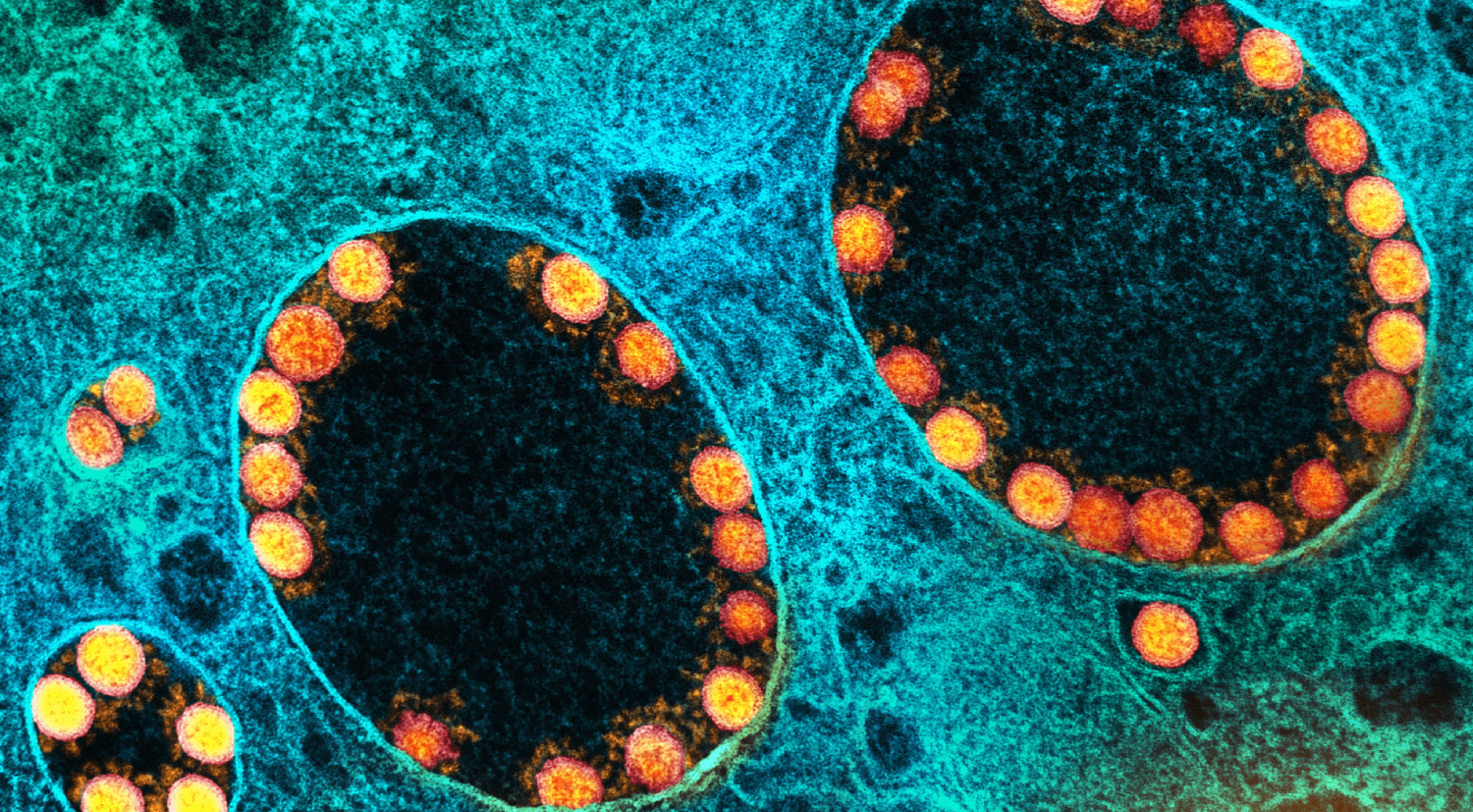
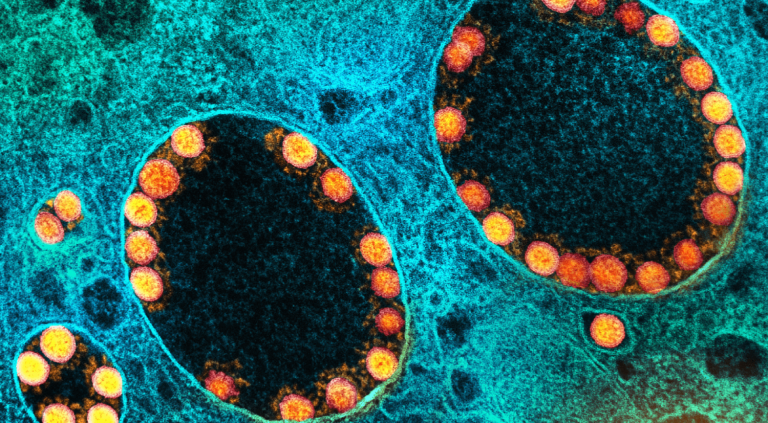
Image d’en tête : Micrographie électronique à transmission de particules du virus SARS-CoV-2 (or) dans les endosomes d’une cellule épithéliale olfactive nasale fortement infectée. Image prise au NIAID Integrated Research Facility (IRF) à Fort Detrick, Maryland. CC by-sa 2,0 NIAID, via Flickr
En ce cinquième anniversaire du confinement en France, cet article propose une synthèse des données scientifiques et autres révélations postérieures à juillet 2021 sur l’origine et les répercussions du Covid 19, faisant suite aux précédents articles écrits avant cette date par les mêmes auteurs.
Cet article vient compléter les 3 articles déjà parus dans Parlons Science. Il fait un état des lieux des variants et des vaccins disponibles à ce jour et se penche sur l’origine du virus SARS-CoV-2 qu’il parait indispensable d’élucider afin d’éviter de futures crises sanitaires mondiales.
Le virus SARS-CoV-2 s’est répandu très rapidement sur notre planète depuis la fin de l’année 2019. Responsable des symptômes associés à la maladie appelée Covid-19, ce virus a complètement bouleversé nos vies et la plupart de nos sociétés.
Un nouveau virus mortel vient de faire son apparition en Chine. A ce jour, plus de 2 000 personnes ont été infectées depuis le mois de décembre et 80 en sont mortes. 3 cas ont été identifiés en France. Quel est ce virus, d’où provient-il et comment se transmet-il ?
Votre abonnement à la lettre d’information : Muséum de Toulouse