Article rédigé le 10 Juin 2020 parHENRI CAP, Biologiste et zoologue au Muséum de ToulouseDOMINIQUE MORELLO, Biologiste retraitée, CNRS
Après deux premiers articles sur le SARS-CoV-2, le Muséum d’Histoire Naturelle de Toulouse achève son investigation sur l’étude de ce virus. Afin de mieux comprendre son comportement à l’intérieur de nos cellules, du fait de sa morphologie, de son génome et de son cycle de vie particulier, nous nous intéresserons à sa position systématique dans l’histoire de l’évolution, à ses relations de parenté avec les autres virus et nous tenterons d’expliquer son origine, qui reste, encore aujourd’hui, une énigme.
Une origine naturelle ou bien artificielle ?
Deux théories s’affrontent à l’heure actuelle pour tenter de répondre aux questions sur l’origine du SARS-CoV-2. La première, soutenue par une majorité de scientifiques, voudrait que ce virus soit entièrement d’origine naturelle, et que l’intervention humaine se soit limitée à favoriser son émergence. Plusieurs facteurs ont été évoqués comme la déforestation et l’extension des terres agricoles, une promiscuité accrue entre certains animaux et l’homme du fait de la surpopulation ou de conditions de détentions des animaux sauvages issus du trafic dans des marchés insalubres, ou encore du réchauffement climatique.
L’autre hypothèse est soutenue par quelques scientifiques et une partie de la population chinoise et mondiale, dont plusieurs états et médias américains et leur président. Ils estiment que le virus a été créé artificiellement dans un laboratoire de virologie situé à Wuhan en Chine, lieu de départ de l’épidémie. Avant d’aborder cette seconde hypothèse pour le moins sulfureuse, intéressons-nous aux données qui sont à notre disposition pour étayer l’hypothèse naturelle.
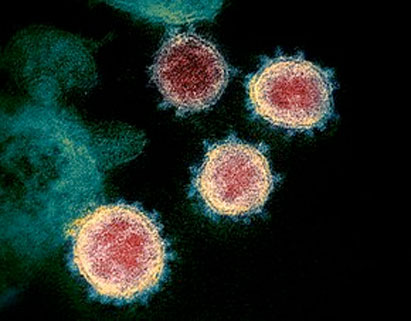
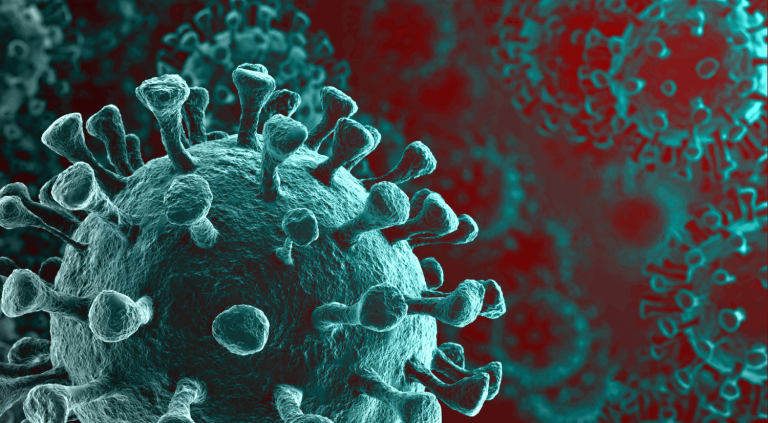
Observation de coronavirus SARS-CoV-2 au microscope électronique. Crédit : NIAID-RML
Hypothèse naturelle
Regardons tout d’abord les données de séquençage des génomes de différents coronavirus pour connaître les relations de parentés du SARS-CoV-2 avec les autres coronavirus qui parasitent les mammifères et les oiseaux depuis des millions d’années.
Position systématique
Au sein des coronavirus, sept sont spécifiques à l’espèce humaine (229E, NL63, OC43, HKU1, MERS-CoV, SARS-CoV-1 et SARS-CoV-2). Si les quatre premiers n’engendrent généralement que de simples rhumes, les trois autres ont entraîné des syndromes respiratoires graves (cf articles 1 et 2). Tous appartiennent à la sous-famille des Orthocoronavirinés. Parmi ces derniers, le SARS-CoV-2 est classé dans le genre Betacoronavirus, caractérisé par la présence à sa surface de la protéine hémagglutinine estérase (HE), et le sous-genre Sarbecovirus qui ne présente qu’une seule protéase (enzyme qui coupe les protéines) de type papaïne (PLpro). Les hôtes naturels des Sarbecovirus sont principalement les chauve-souris (Bat).
Premières phylogénies : Les chauves-souris rhinolophes réservoirs naturels des coronavirus
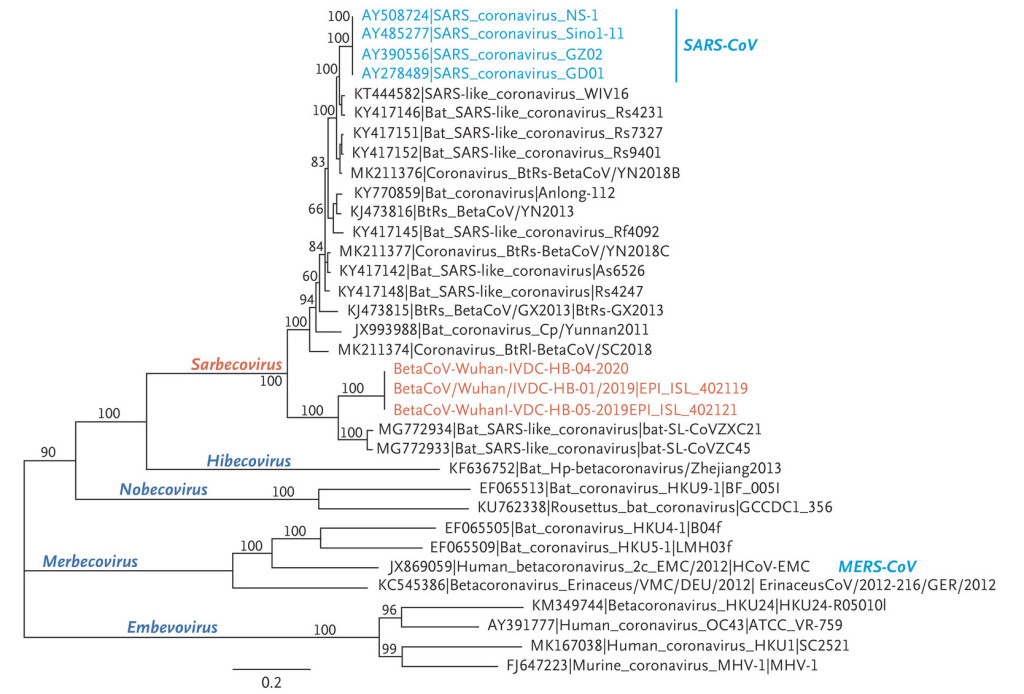
Première analyse phylogénétique du génome du virus SARS-CoV-2 (en orange) et d’autres Betacoronavirus. Credit : Zhu et al. 2020
Une première comparaison d’un panel d’échantillons de SARS-CoV-2 prélevés chez des malades avec les génomes d’autres Betacoronavirus a montré qu’il ne provient pas du SARS-CoV-1 responsable de la première épidémie de SRAS en 2002, car ils n’ont que 82 % de séquences communes, laissant supposer que ces deux virus appartiennent à deux lignées divergentes qui se sont séparées il y a plusieurs décennies. Le SARS-CoV-2 serait plus proche, à 89%, du coronavirus (ZC45) de la chauve-souris rousse chinoise en Fer à cheval, Rhinolophus sinicus, une espèce de rhinolophe (Zhu et al. 2020). Cependant, en mars, une deuxième analyse effectuée à partir d’une autre population de chauve-souris, montre une parenté encore plus proche avec un coronavirus prélevé sur une autre espèce de rhinolophe, Rhinolophus affinis, appelé le Fer à cheval intermédiaire (Hassanin 2020). Ce virus baptisé RaTG13 présente une séquence génétique globale identique à 96% avec le virus SARS CoV-2 (Zhou et al. 2020).
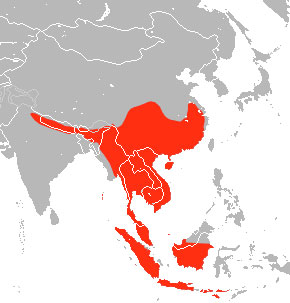
Rhinolophe Fer à cheval intermédiaire en position de repos (Credit: Andrew Pierce) et son aire de répartition naturelle (Crédit : Wikipédia).
Ce résultat confirme bien lerôle des chauves-souris comme réservoir naturel de nouveaux virus. A l’instar d’autres animaux sauvages, ces dernières hébergent des virus, sans en être affectées outre mesure (Leroy 2020), et peuvent les transmettre à d’autres espèces, si la barrière spécifique est franchie lors de rapprochements fortuits ou imposés (prédation, trafic et marchés alimentaires). Historiquement, ce scénario s’est probablement déjà produit lors des deux épidémies précédentes du SRAS de 2002 et du MERS de 2012, mais à chaque fois, le virus provenant des chauves-souris semble avoir été transmis à une espèce intermédiaire, en l’occurrence le dromadaire, Camelus dromedarius, pour le MERS et la civette masquée, Paguma larvata, pour le SRAS. Le SARS-CoV-1 a, en effet, 99.8% de son génome homologue avec celui du coronavirus de la civette masquée (Hassanin 2020). Ainsi, l’infection de ces espèces intermédiaires pourrait favoriser le passage ultérieur du virus à l’humain comme cela s’est déjà produit en Australie en 1994 avec le virus Hendra, dont la contagion des chevaux a gagné l’homme, puis en Malaisie en 1998 avec le virus Nipah, qui passa des porcs aux êtres humains. A chaque fois, l’épidémie était causée par des virus de grandes chauve-souris mangeuses de fruits, les chevaux et les porcs étant les hôtes intermédiaires de la contamination de l’homme (Wang et Eaton 2007).
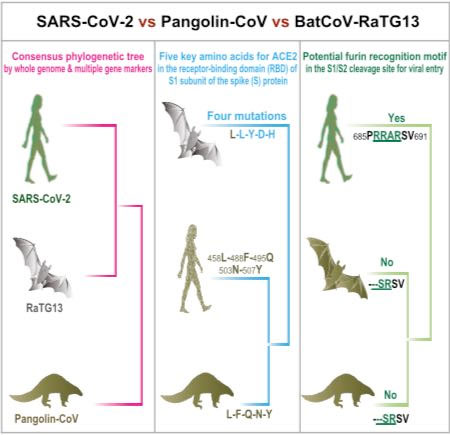
Dans le cas du SARS-CoV-2, il se pourrait qu’il y ait aussi un hôte intermédiaire. Voyons pourquoi. Le virus porte à sa surface une protéine capitale, la protéine S (Spike en anglais), car elle se lie au récepteur de la membrane des cellules de l’hôte auquel elle est adaptée, comme une clé dans une serrure, l’enzyme de conversion de l’angiotensine 2 (ACE2). Or il se trouve que la partie de la protéine S qui se lie au récepteur ACE2 (le domaine de liaison, Figure 4) n’est homologue qu’à 77% avec le virus du Fer à cheval intermédiaire : RaTG13 (Zhang et al. 2020 ; Hassanin 2020). En d’autres termes, ce coronavirus de chauve-souris ne s’attacherait pas aux cellules humaines de façon optimale, car l’extrémité de sa protéine S ne serait pas parfaitement adaptée au récepteur.
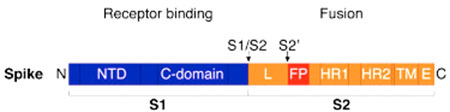
Figure 4. Schéma de la protéine S et son domaine de liaison au récepteur (Receptor binding, S1 à gauche) et le domaine de fusion à la membrane (S2 à droite). S1/S2 et S2’ représentent les principaux sites de clivage. Crédit : Millet et Whittaker 2015
Le pangolin malais : hôte intermédiaire pas si idéal
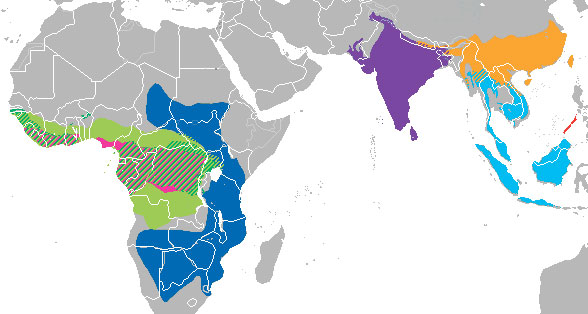
En Février, la revue Sciences et Avenir, ainsi que l’ensemble des médias, relaye un communiqué de l’Université du Sud de la Chine déclarant qu’après avoir testé plus de 1000 échantillons d’animaux sauvages, des chercheurs chinois ont montré que les génomes de coronavirus prélevés sur unpangolin malais étaient à 99% identiques à ceux trouvés chez les patients atteints du Covid-19. Rappelons que les pangolins sont des mammifères qui se situent phylogénétiquement entre les chauves-souris et les carnivores, et qu’il existe 8 espèces réparties équitablement en Afrique et en Asie (Figure 5). Toutes les espèces font l’objet d’un immense trafic illégal, car les pangolins constituent la viande de brousse la plus exploitée au monde avec 100 000 individus abattus chaque année. Le pangolin malais ou javanais, Manis javanica, est de ce fait en danger critique d’extinction sur son aire de répartition, du sud de la Chine au sud du Yunnan, à la Malaisie et à l’Indonésie (Hassanin 2020).
Figure 5. Pangolin malais en position d’escalade et son aire de répartition naturelle (en bleu clair) comparée à celle des autres espèces de pangolins. Crédit : Wikipédia
Ces informations se sont finalement révélées fausses car les génomes des coronavirus isolés chez les pangolins présentent entre 86 et 92 % de similarité avec le SARS-CoV-2, ce qui suggère une séparation il y a plusieurs décennies. En revanche, pour un isolat particulier, la souche de Guangdong, l’homologie est de 97,4% dans la partie de la protéine S qui correspond au fameux domaine de liaison au récepteur ACE2 (d’une longueur de 74 acides aminés). Comme nous l’avons vu plus haut, c’est bien supérieur au 77% du virus RaTG13 pour la même région, suggérant que ce virus aurait pu contaminer un être humain, une hypothèse que propose Alexandre Hassanin, spécialiste en phylogénie moléculaire au MNHN (Hassanin 2020).
Toutefois, il subsiste un autre problème de taille que nous allons analyser : Une fois que le virus s’est attaché au récepteur ACE2, son enveloppe fusionne avec la membrane de la cellule hôte . Mais cette étape nécessite de rendre accessible le domaine de fusion de la protéine S (Figure 4) ce qui n’est possible qu’après coupure (ou clivage) de cette très grosse protéine de plus de 1200 acides aminés. Plusieurs sites de clivage ont été décrits (voir Figure 4) qui sont potentiellement reconnus par plusieurs enzymes de la cellule hôte dont la furine qui coupe au site S1/S2. C’est unespécificité du SARS-CoV-2 au sein des Sarbecovirus, même sion peut retrouver ce site chez d’autres coronavirus moins apparentés, tel que le MERS-CoV.
Or, pour en revenir au pangolin, aucune des séquences de coronavirus de pangolins analysées jusqu’à présent ne comporte le site de clivage S1/S2 adapté à la furine (Figure 6).
Figure 6. Relations de parenté du SARS-CoV-2 avec les virus du rhinolophe intermédiaire RaTG13 et du pangolin selon leur génome (rose), le domaine de liaison au récepteur ACE2 (bleu), le site de clivage furine S1/S2 (vert). Crédit : Zhang et al. 2020
Mutations et recombinaisons pour expliquer l’inexplicable
Mais alors ? Des scientifiques comme Kristian Andersen du département d’immunologie et de microbiologie de l’Institut de Recherche SCRIPPS à La Jolla aux Etats-Unis proposent un scénario évolutif reposant sur une double sélection naturelle, une ayant eu lieu dans l’hôte intermédiaire, puis l’autre chez l’humain. Au fil du temps et des mutations, des virus possédant la protéine S du SARS-CoV-2 avec un domaine de liaison adapté au mieux au récepteur ACE2 des cellules humaines auraient été sélectionnés dans l’hôte intermédiaire, qui pourrait être le pangolin ou un autre hôte intermédiaire à découvrir ; puis des mutations se seraient produites dans les cellules humaines, aboutissant après de multiples transmission inter-humaines silencieuses, à la sélection d’une souche virale ayant dans sa protéine S des sites de clivage adaptés aux protéases cellulaires, telle la furine qui favoriserait la fusion du virus aux cellules humaines. La question que l’on peut se poser, c’est comment ce virus aurait développé une protéine S adaptée aux cellules humaines dans les cellules de l’hôte intermédiaire ?
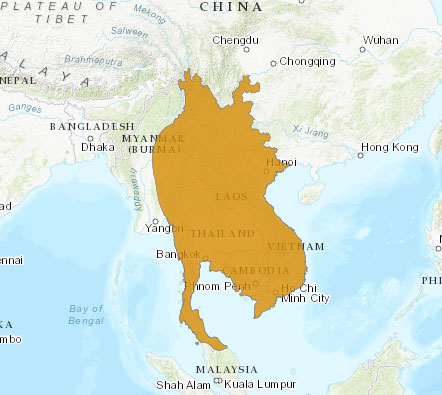
Depuis cette hypothèse, une nouvelle analyse génétique de 227 chauves-souris appartenant à 20 espèces différentes du Sud de la Chine révèle que la chauve-souris malaise en fer à cheval, Rhinolophus malayanus (Figure 7) héberge un virus, au doux nom de RmYN02, dont le génome présente globalement 93,3% d’homologie avec le SARS-CoV 2 et qui possède une séquence de clivage S1/S2.
Figure 7. Chauve-souris malaise en fer à cheval, Rhinolophus malayanus, (Crédit : mammalwatching.com) et son aire de répartition. Crédit : UICN
La partie n’est pas pour autant gagnée avec cette nouvelle publication car ce site de clivage ne correspond pas exactement à celui du SARS-CoV-2. Il ne peut donc pas être reconnu par la furine et n’est de ce fait pas fonctionnel. De plus, globalement les séquences de la protéine S de RmYN02 et de son domaine de liaison au récepteur ne sont respectivement que de 71,9% et 61,3% homologues à celles du SARS-CoV-2, ne permettant pas une liaison optimale au récepteur ACE2.
Si cette piste est la bonne, il faut postuler que l’un ou l’autre de ces coronavirus de chauve-souris se soient recombinés avec le virus d’un hôte intermédiaire qui reste à trouver, ou qu’il y a eu une transmission à cet hôte d’un virus qui aurait acquis par dérive de mutations sélectionnées les capacités à infecter efficacement l’espèce humaine.
Dans les deux cas, cela impliquerait une contamination naturelle entre une chauve-souris et un hôte intermédiaire dans leur milieu d’origine en Asie du Sud-Est. Comme le souligne Alexandre Hassanin, il est peu probable que le pangolin malais soit cet hôte intermédiaire car, contrairement aux chauve-souris dont le système immunitaire est très efficace, il semble très sensible au virus, limitant sa diffusion. D’autre part, il est très rare, classé en danger critique d’extinction, et solitaire ce qui limite les contacts éventuels. On peut davantage envisager une contamination des pangolins lors de leur captivité dans des cages, durant leur transport ou dans des marchés de faune sauvage. Cette hypothèse serait étayée par l’exemple de la civette incriminée dans l’épidémie de SARS-CoV-1 : la recherche du virus sur plus de 1100 civettes échantillonnées dans des fermes d’élevage s’est révélée négative tandis que les 6 civettes testées dans un restaurant de Guangzhou fréquenté par des personnes infectées étaient porteuses d’un virus à 99,8% identique à celui du SARS-CoV-1. Il serait donc probable qu’elles aient été contaminées lors de leur captivité en raison de leur promiscuité avec d’autres animaux sauvages (Hassanin 2020), ou qu’elles aient été contaminées par des humains infectés, ce qui changerait beaucoup de choses !
Les conditions de stockage des cages dans des marchés, comme celui de Wuhan suspecté d’être le point de départ de l’épidémie de Covid 19, auraient pu favoriser le rapprochement d’animaux qui dans la nature ne se rencontrent jamais et permettre une contamination interspécifique. En tout état de cause, si le pangolin est une bonne piste comme hôte intermédiaire, il ne peut être à l’origine du virus humain du fait de la trop grande divergence génétiquede son coronavirus avec leSARS-CoV-2 et de l’absence de recombinaison entre son virus et celui d’une chauve-souris (Boni et al. 2020).
Un autre hôte intermédiaire est vivement recherché
Le virus SARS CoV-2 se révèle être un virus ubiquiste qui peut infecter une large gamme d’espèces (Andersen et al. 2020), comme en attestent les nombreux cas de carnivores domestiques atteints par la maladie, essentiellement des chats mais aussi des chiens, furets et tigres, comme celui du zoo de New-York (Brassard 2020). Ainsi les carnivores sauvages vendus sur les marchés chinois, comme les chiens viverrins, Nyctereutes procyonoides, ou plusieurs espèces de blaireaux asiatiques, sont actuellement des espèces sur lesquelles les recherches vont s’intensifier car l’origine naturelle du Covid 19, si elle était confirmée, serait une conséquence directe du trafic illégal de faune sauvage. Les zones de stockage, comme les marchés d’animaux, constituent en effet des aires privilégiées pour la circulation de ce type de virus (Hassanin 2020).
Cependant, si les espèces de carnivores domestiques ont été contaminées de façon certaine par l’homme, pourquoi ne serait-ce pas également le cas pour les espèces hôtes considérées comme intermédiaires ? En cela, certains travaux déjà publiés mériteraient d’être ré-analysés selon John Wenzel, spécialiste de systématique phylogénétique du Carnegie Museum de Pittsburgh (Wenzel 2002), qui rappelle dans un article à paraître dans le bulletin de la Société Française de Systématique (https://sfs.snv.jussieu.fr/) que l’hypothèse considérant la civette masquée comme hôte intermédiaire dans la première épidémie de SRAS de 2002 a été invalidée, suite aux nouvelles analyses phylogénétiques effectuées à partir des séquences disponibles sur les plate-formes internationales telles que GenBank. Les résultats sont sans équivoque : « Quels que soient les paramètres d’alignement, les critères d’optimalité ou l’échantillonnage isolé,les phylogénies qui en résultent montrent clairement que le SARS-CoV-1 a été transmis aux petits carnivores bien après l’épidémie de SRAS chez l’homme, qui a débuté fin 2002 » (Janies 2019).
Dans le cas du SARS-CoV-2, plusieurs problèmes méthodologiques ont été soulevés en particulier dans les analyses phylogénétiques qui manquent, pour certaines, d’une racine valable pour établir des relations de parenté ; la taille des échantillons n’est pas toujours suffisante pour tirer des conclusions ; des prélèvements sur d’autres isolats provenant d’espèces hôtes de différentes sortes sont aussi indispensables pour conclure avec certitude que la source ancestrale probable est bien celle à laquelle on pense (Wenzel 2020).
Hypothèse artificielle
Figure 8.Représentation artistique du SARS-CoV-2 et de son origine, un virus de chauve-souris. Crédit : Richard Borge/ Scientific American (2020) 322, 6, 24-32)
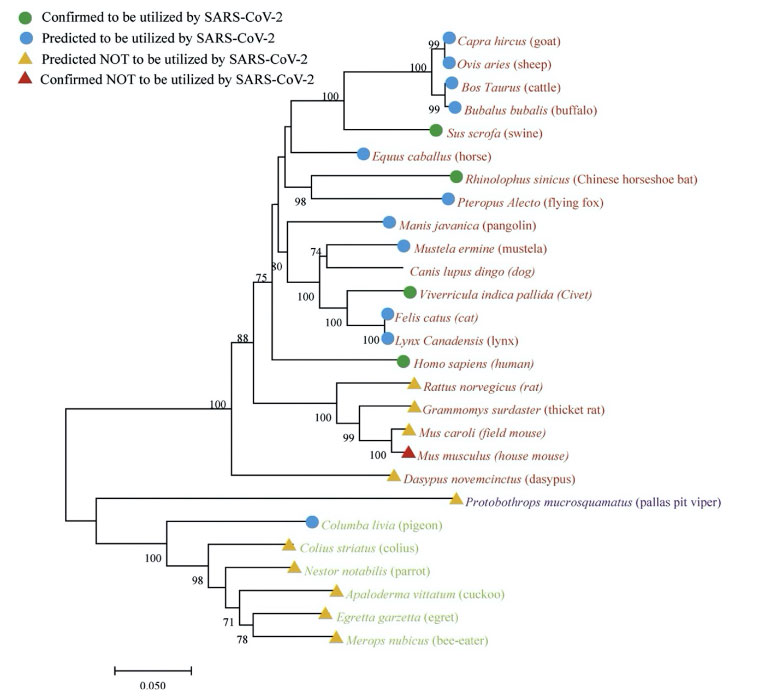
L’ensemble des publications privilégiant l’hypothèse naturelle du SARS-CoV-2 ne nous permet pas en l’état actuel de nos connaissances d’écarter l’hypothèse d’une création artificielle dans un laboratoire. Une des raisons principales est que le SARS-CoV-2 se fixe et pénètre plus facilement les cellules humaines que celles de chauve-souris, alors que ces animaux sont censés en être la source ! (Piplani et al. 2020).
Figure 9.Relations phylogénétiques des récepteurs ACE2 de différentes espèces dont l’homme (Homo sapiens) et la chauve-souris rousse chinoise en Fer à cheval (Rhinolophus sinicus) établies à partir de leurs séquences homologues, et leur utilisation par la protéine S du virus SARS-CoV-2. Crédit : Qiu et al 2020, Piplani et al. 2020
Les scientifiques savent recréer artificiellement un virus déjà connu, comme cela a été fait officiellement pour la première fois en 2003 avec le virus bactériophage PhiX174 (un virus qui infecte une bactérie), à partir d’oligonucléotides de synthèses (Smith et al. 2003). De plus, avec les outils puissants de biologie moléculaire tels que Crispr-Cas9, il est possible de fabriquer de nouveaux virus de toutes pièces, à partir de virus déjà connus et conservés dans les bibliothèques d’échantillons d’un laboratoire. A ce titre, le laboratoire de Wuhan (Wuhan Institute of Virology (WIV), qui se trouve être le point de départ de l’épidémie, est spécialisé dans l’étude des coronavirus de chauve-souris. Il inclut un laboratoire de haute sécurité dit « P4 », construit avec l’aide de plusieurs pays dont la France et les Etats-Unis, inauguré en 2017, qui lui permet de répondre à son objectif principal : étudier l’évolution des virus lors des transmissions entre animaux, et repérer l’émergence de mutations potentiellement dangereuses pour l’humain (Hecketsweiler 2020).
Plusieurs publications scientifiques montrent que des équipes chinoises menées par Zhenghli Shi, la directrice de ce laboratoire, en collaboration avec des équipes étrangères, notamment des laboratoires américains, manipulaient des virus de chauve-souris pour les rendre plus infectieux (Menachery et al. 2015). Etienne Decroly, spécialiste des virus émergents au CNRS considère que ces travaux constituent « un risque important, notamment en cas de contamination accidentelle. D’autre part, le seul fait de cultiver des virus dans des cellules humaines ou de primates soulève des questions, car avec le temps, ils peuvent s’adapter, et acquérir un pouvoir pathogène pour l’homme qu’ils n’avaient pas« . Il suggère ainsi qu’ « on ne peut pas écarter l’hypothèse que le SARS-CoV-2 provienne de leur collection et se soit échappéà la suite d’une contamination accidentelle, mais, à moins d’avoir accèsà leurs cahiers de laboratoire, on n’en saura jamais rien » (Hecketsweiler 2020), d’autant que le laboratoire de Wuhan reste fermé aux observateurs étrangers.
Dans leur article de 2015, Menachery et ses collaborateurs expliquent justement comment la création de virus chimères à partir de souches naturelles pour augmenter leurs capacités de contamination et de pathogénicité sur l’homme pourrait aboutir à une pandémie comme celle du SARS-CoV-2 (Menachery et al. 2015).
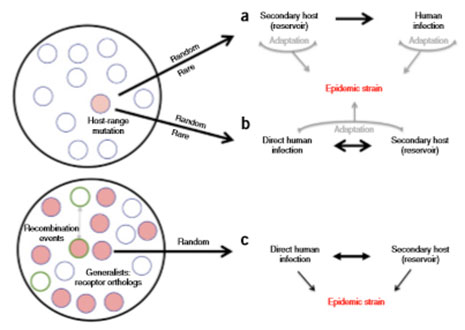
Figure 10. Les trois paradigmes d’émergence des coronavirus pouvant aboutir à des épidémies. Crédit : Menachery et al. 2015
D’après les auteurs, des coronavirus pathogènes peuvent émerger en suivant trois paradigmes : les deux premiers stipulent que des virus mutants (en rouge sur le schéma 10) peuvent apparaître par hasard mais rarement au sein de leur espèce hôte. Leurs mutations ponctuelles rares leur permettent de s’adapter soit à une espèce hôte secondaire (a), facilitant ainsi son passage à l’homme, soit directement à l’homme (et éventuellement par la suite une autre espèce hôte secondaire) (b). Le dernier paradigme repose sur la création de virus chimères « augmentés », résultant de recombinaisons entre virus aux capacités complémentaires, infectant l’homme ou un hôte secondaire, et déclenchant ainsi une épidémie (c). On se demande en lisant cet article si le SARS-CoV-2 ne serait pas tout droit sorti de cette étude… D’autant qu’une pandémie accidentelle s’est déjà produite en 1977, avec le virus de la grippe russe, qui était quasiment identique au virus H1N1 de la grippe espagnole qui était en circulation dans les années 1950. Conservé dans des fioles en laboratoire, ce virus serait d’abord apparu en Chine.
Le marché et les jeux mondiaux militaires de Wuhan
Plusieurs autres arguments pourraient également étayer l’hypothèse artificielle du SARS-CoV-2. La première concerne la piste du marché de Wuhan, un temps suspecté d’être le point de départ de l’épidémie. Elle se révèle infructueuse d’après l’identification du premier cas recensé qui n’aurait pas fréquenté le marché (Huang et al. 2020). D’autre part, il est avéré que ce marché n’a jamais vendu de chauve-souris car la population locale n’en consomme pas, mise à part quelques grandes chauves-souris ou roussettes, mais aucun micro-chiroptères de type rhinolophes, dont les coronavirus sont les plus proches génétiquement du SARS-CoV-2. En revanche, l’hôte intermédiaire suspecté, le pangolin malais, a pu s’y retrouver avant ou pendant son interdiction de chasse et de commerce. Comme les registres du marché ont été réquisitionnés par les autorités chinoises, il n’y a aucun moyen de le vérifier.
De plus, si l’épidémie a débuté officiellement à la fin du mois de décembre avec un premier foyer de contaminations localisé au marché de Wuhan, comme l’ont signalé les médecins lanceurs d’alertes chinois dont on a perdu la trace, le cas le plus ancien remonterait en fait un mois et demi avant, au 17 novembre, selon le quotidien South China Morning Post, qui s’appuie sur des données gouvernementales non publiées. Cette même date est aussi avancée par Andrew Rambaut, de l’Université d’Edimbourg, qui conclut après analyse des différentes souches et mutations du virus à un ancêtre commun datant du même 17 novembre (Andersen et al. 2020). A ce jour, les génomes de près de 20 000 virus SARS-CoV-2 ont été séquencés, permettant de tracer sa circulation grâce à son évolution génétique, et ces résultats ne concordent pas avec la version officielle du régime chinois.
L’autre événement troublant se situe le 18 octobre, soit un mois avant le premier cas estimé et plus de deux mois avant le premier cas officiel. Il s’agit des jeux mondiaux militaires de Wuhan. De façon réciproque et surprenante, la Chine renvoie la paternité supposée d’une origine artificielle du virus aux Etats-Unis, les accusant d’avoir amené le virus avec eux à l’occasion de ces jeux mondiaux et de l’avoir disséminé dans les équipes de militaires venant du monde entier. Près de 10 000 participants étaient logés au même endroit ; et après leur retour, de nombreux cas de maladies suspectes ont été rapportés au sein de la délégation française ou italienne. Sur place à Wuhan, cinq militaires sportifs ont été admis dans des hôpitaux pour des crises de malaria selon les autorités chinoises. A ce petit jeu de déclarations au cours duquel le président des Etats-unis a surenchéri en accusant à son tour la Chine, ces deux super puissances semblent renvoyées à leurs responsabilités (Guibert et al. 2020). Quoiqu’il en soit, de quel virus les athlètes étaient-ils porteurs, d’un virus « naturel » ou d’un virus chimère fabriqué en collaboration avec des scientifiques chinois, qui se serait échappé d’un laboratoire américain ou chinois…?
A quand des certitudes ?
Pour l’instant, bien que de nombreuses interrogations subsistent, la communauté scientifique continue de pencher pour l’hypothèse naturelle de l’émergence du SARS-CoV-2. La recherche dans ce domaine est en pleine ébullition et des résultats d’études en cours sont attendus avec impatience pour expliquer ce qui reste à ce jour inexplicable.
Et après demain ?
Si la pandémie du Covid-19 a durement frappé les plus fragiles, que ce soit les malades ou ceux qui ont perdu leur emploi suite au confinement, elle a également révélé des vertus positives que chacune et chacun d’entre nous ont pu constater. Le dévouement du personnel soignant des hôpitaux avec les risques que cela comportait (catégorie professionnelle la plus contaminée), tout comme le maintien de l’approvisionnement alimentaire du fait de la présence d’hommes et surtout de femmes qui en assuraient le fonctionnement, sans parler de l’entraide, tout ceci nous a montré la plus belle facette de l’être humain, un animal empathique à l’extrême prêt à tout pour sauver des vies.
En même temps, l’arrêt brutal de l’activité humaine a constitué pour la nature un don du ciel. Le fait que tout s’arrête dans les villes du monde entier a donné l’occasion à certaines espèces de se réapproprier le territoire que nous occupons sans partage en s’exprimant à nouveau, et cela a fait du bien d’entendre les oiseaux chanter. Espérons que le dé-confinement et la reprise économique n’effacent pas ces effets bénéfiques, inespérés. Alors quelle que soit l’origine du Covid-19, des mesures pourraient être prises pour minimiser les chances qu’une telle pandémie se reproduise, naturellement ou pas.
Les mesures qui pourraient être prises
En premier lieu, afin de limiter les risques naturels, il s’agirait enfin d’appliquer les lois sur la protection des espèces sauvages menacées, en interdisant définitivement le commerce de celles qui sont à l’origine de zoonoses (Hassanin 2020). D’autre part, la déforestation et le développement des activités agricoles et économiques à outrance doivent être davantage régulés pour ne pas favoriser l’émergence de virus à partir d’espèces qui n’avaient auparavant pas de contact avec l’homme (Leroy 2020).
Un des objectifs du projet de recherche Discovery, financé par l’Agence nationale de recherche, vise à mieux comprendre l’émergence des virus et à cibler les pratiques à risque, tel que l’élevage d’hôtes potentiellement intermédiaires ou la capture d’espèces réservoirs comme les chauve-souris. Car le lien entre la santé des écosystèmes et la santé humaine est aujourd’hui clairement établi (Grandcolas et Justine 2020).
En parallèle, la re-localisation des sites de productionde matériels liés à la santé (masques, matériel médical et médicaments) semble vitale, tout comme l’augmentation des capacités en lits de réanimations des hôpitaux et du personnel qui y travaille sans compter, avec les risques que cela comporte.
Enfin, notre impact moindre sur un territoire donné pendant le confinement, notamment dans les villes, a non seulement fait baisser le niveau de pollution de l’air et des rivières, et celui du réchauffement de l’air induit par nos activités, mais a également sauvé la vie de milliards d’insectes ; un effet bénéfique pour les plantes, car leur reproduction a été favorisée, mais aussi pour les animaux qui ont trouvé davantage de nourriture. En ce sens, toute minimisation de notre impact sur l’environnement représente une mesure profitable à toute la planète.
Dans un second temps, il est nécessaire de prendre conscience de nos actes. Car depuis des milliers d’années, l’être humain a étendu son emprise sur le territoire des espèces sauvages, réduisant leur espace vital à une peau de chagrin, voire à une cage. Alors que la nature est merveilleuse pour qui sait encore la regarder et s’émerveiller. Nous faisons partie d’elle, comme les deux faces d’un même processus qui se spécifient mutuellement, et si la nature peut être terrible, nous le sommes tout autant par nos actions qui ont pu engendrer ce virus, et par nos réactions excessives face à cette épidémie. Nous avons vu que le hasard des mutations avait peut-être créé ce fléau, à partir de virus pourtant anodins, que l’action de l’être humain sur ces phénomènes naturels pouvait les amplifier de façon involontaire ou délibérée, que sa réaction, à l’instar de son système immunitaire, pouvait les aggraver.
Il devient donc urgent d’organiser un moratoire autour de la recherche sur les pathogènes, et notamment les virus, conservés ou (re)créés dans les laboratoires de haute sécurité, dans certains secteurs de la génétique et du médical. Plusieurs exemples de fabrication de virus mutants ont défrayé la chronique récemment, comme celui du virus de la grippe aviaire H5N1 publiés dans la revue Science en 2011 par le virologue hollandais Ron Fouchier et son équipe. Une commission internationale pourrait ainsi effectuer des contrôles dans n’importe quel pays, afin de vérifier que les recherches sur les vaccins ou sur l’émergence de nouveaux virus, ne puisse aboutir de façon détournée ou à l’occasion d’un changement de régime, à la création d’armes biologiques.
Conclusions
Malgré tout ce qui a été dit, il est important de rappeler qu’une majeure partie de la communauté scientifique ne reconnaît toujours pas les virus comme faisant partie du vivant, du fait qu’ils ne sont pas autonomes pour se reproduire. Pourtant, nous avons vu que leur capacité à s’adapter à leur hôte dépendait autant du hasard, que de leur génome ou de celui de leur hôte, abritant parfois d’autres virus. L’ensemble formant un tout, que nous pourrions appeler vivant, et qui se caractériserait par la possession d’un patrimoine génétique commun : l’ADN ou l’ARN.
C’est le choix qui a été fait au Muséum de Toulouse en présentant ces deux conceptions du vivant dans son exposition permanente. Et si les virus sont vivants, cela pose bien évidemment un problème majeur que de pouvoir favoriser l’émergence d’êtres vivants « augmentés » dans leur capacité à parasiter d’autres êtres vivants, en l’occurrence notre propre espèce ! En créant de nouveaux virus, l’homme s’est octroyé le droit de manipuler la vie, et en cela l’être humain s’est donné beaucoup plus de pouvoir que de sagesse, alors que sa survie ne tient qu’à celle des autres êtres vivants.
D’ailleurs, les virus n’amènent pas que la mort ou la peur, qui ont été martelée par les médias, ils donnent également leurs gènes à leurs hôtes régulièrement : l’ensemble de notre ADN en serait constituée à plus de 10 %.
Ainsi depuis l’apparition de la vie, les virus ont joué un rôle majeur dans l’évolution des espèces, et pour preuve, c’est à l’un d’entre eux que nous devons le fait de ne plus pondre des oeufs, nous ayant donné le gène responsable de la fabrication d’un placenta, il y a plus de 100 millions d’années.
Alors qui sait ce que ce nouveau virus nous léguera, et ce que nous ferons de ce monde, car outre l’origine mystérieuse du SARS-CoV-2, ce qui se passe actuellement en France et dans le reste du monde interroge, comme l’a si bien dit la philosophe et historienne des sciences françaises, Bernadette Bensaude-Vincent, au centre INRAE Occitanie de Toulouse ce 23 Avril: « Alors que ces dernières années la situation de la recherche scientifique n’était guère flamboyante, puisqu’il y a eu des décennies de désengagement de l’Etat, qui a autonomisé les universités, donné plus de place à la recherche privé, or voici que d’un seul coup, mi-mars, plein feu sur la médecine et la science, et toute la science est dans l’horizon d’attente de tests, de vaccins, et donc, on attend de nouveau des promesses de la science, on attend de nouveau des miracles de la science. Serait-on en train de redécouvrir l’utilité de la recherche scientifique ?… » (https://www.youtube.com/watch?v=gxCtsMtiBvc&feature=youtu.be)
Références
Andersen K.G., Rambaut A., Lipkin W.A, Holmes E.C. et R.F. Garry. 2020. « The proximal origin of SARS-CoV-2 », nature medecine, no 26, 17 mars 2020, p. 450-452
Boni M., Lemey P., Jiang X., Lam T.T.Y., Perry B., Castoe T., Rambaut A., Robertson D.L. 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. bioRxiv doi.org/10.1101/2020.03.30.015008
Bonnin A. 2018. Caractérisation de la protéine S du coronavirus humain 229E. Thèse de doctorat. Ecole doctorale Biologie-Santé (Lille).
Brassard C. 2020. Et si mon animal était contaminé ? Le point sur les chats, les chiens et les furets. The Conversation 7 Avril 2020
Campan R. et F. Scapini. Ethologie approche systémique du comportement animal. De Boeck.
Coutard B., Valle C., de Lamballerie X. et B. Canard, « The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade », Antiviral Research, vol. 176, avril 2020, p. 104742
Crosnier C., Bustamante, L.Y, Bartholdson S.J., Bei, A.K. 2011. « Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum », Nature, 480, no 7378
Cyranoski D. 2020. Profile of a killer virus. Nature, 581, 22-26.
Ge X.Y., Li J.L., Yang Y.L., et al. 2013. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature, 503, 535-538.
Gheblawi et al. 2020. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circulation Research, 126, 1456-1474.
Grandcolas P. et Justine D.L. 2020. Covid-19 ou la pandémie d’une biodiversité maltraitée. The Conversation 25 Mars 2020.
Guibert N., Thibault H., et C. Guillou. 2020. Ce que l’on sait des Jeux mondiaux militaires de Wuhan, après lesquels plusieurs athlètes disent être tombés malades. Le Monde le 12 mai 2020.
Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P. H., Zhang L.J., Guan Y.J. et al. 2003. Isolation and Characterization of Viruses Related to the SARS Coronavirus From Animals in Southern China. Science 302, 5643, 276-278.
Hassanin A. 2020. Cov-19 : Origine naturelle ou anthropique? The conversation. 15 Avril 2020.
Hecketsweiler C. 2020. Coronavirus : le SARS-CoV-2 est-il sorti d’un laboratoire ? Le Monde 17 avril 2020.
Hoffmann M., Kleine-Weber H., Schroeder S., et N. Krüger. 2020. « SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor », Cell, mars 2020
Huang C. et al. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497-506
Janies D. 2019. Phylogenetic Concepts and Tools Applied to Epidemiologic Investigations of Infectious Diseases. Microbiol Spectrum 7(4):AME-0006-2018. doi:10.1128/microbiolspec.AME-0006-2018.
Leroy E. 2020. Les chauves-souris, source inépuisable de virus dangereux pour les humains ? The Conversation 30 Mars 2020.
Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri M. Hu Z., Zhang H., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. 2005. Bats Are Natural Reservoirs of SARS-Like Coronaviruses.Science 310, 5748, 676-679.
Menachery et al. 2015. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nature medecine 21, 1508-1513.
Millet K.J. etWhittaker G.R. 2015. Host Cell Proteases: Critical Determinants of Coronavirus Tropism and Pathogenesis, Virus Res. Avril 2015 https://pubmed.ncbi.nlm.nih.gov/25445340/
Piplani S., Singh P.K., Winkler D.A., Petrovsky N. 2020. N silico comparison of spike protein-ACE2 binding affinities across species; significance for the possible origin of the SARS-CoV-2 virus. arxiv.org/abs/2005.06199arXiv:2005.06199v1.
Qiu Y. et al. 2020. Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptorof SARS-CoV-2. Microbes and infections, doi:10.1016/j.micinf.2020.03.003.
Smith H.O., et al. 2003. Generating a synthetic genome by whole genome assembly: {phi}X174 bacteriophage from synthetic oligonucleotides. », Proc. Natl. Acad. Sci. USA, 100, 15440-15445.
Walls A.C., et al., 2020, Structure, function and antigenicity of the Sars cov 2 spike glycoprotein. Cell 180, 281-292.
Wang L.F., et Eaton B.T. 2007. Bats, Civets and the Emergence of SARS. In: Childs J.E., Mackenzie J.S., Richt J.A. (eds) Wildlife and Emerging Zoonotic Diseases: The Biology, Circumstances and Consequences of Cross-Species Transmission. Current Topics in Microbiology and Immunology, p 325-344. Springer, Berlin, Heidelberg.
Wenzel, J. W. 2002. Phylogenetic analysis: The basic method. Pages 4-30, in R. DeSalle, G. Giribet, and W.Wheeler, eds., Techniques in Molecular Systematics, Birkhäuser.
Wenzel J. 2020. Le SRAS et le COVID-19 n’ont pas de lien avec les civettes et les pangolins. Bulletin de la Société Française de Systématique, sous presse.
Zhang T, Wu K.F., Zhang Z. 2020. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Current Biology 30, 1346-1351.
Zhou, H., Chen, X., Hu, T., et al. 2020. A novel bat coronavirus reveals natural insertions at the S1/S2 cleavage site of the Spike protein and a possible recombinant origin of HCoV-19. bioRxiv 10.1101/2020.03.02.974139.
Zhu, N., Zhang, D., Wang, W et al. 2020 A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med, Février 2020.
Remerciements à Mickael Cohen du CDG31 pour la majeure partie de la documentation scientifique de cet article qu’il a transmis aux auteurs de façon régulière, à Pierre Deleporte, chercheur retraité du CNRS (Université de Rennes 1, EthoS) et à John Wenzel du Carnegie Museum of Natural History de Pittsburgh pour leur relecture bienveillante.
En ce cinquième anniversaire du confinement en France, cet article propose une synthèse des données scientifiques et autres révélations postérieures à juillet 2021 sur l’origine et les répercussions du Covid 19, faisant suite aux précédents articles écrits avant cette date par les mêmes auteurs.
Cet article vient compléter les 3 articles déjà parus dans Parlons Science. Il fait un état des lieux des variants et des vaccins disponibles à ce jour et se penche sur l’origine du virus SARS-CoV-2 qu’il parait indispensable d’élucider afin d’éviter de futures crises sanitaires mondiales.
Le virus SARS-CoV-2 s’est répandu très rapidement sur notre planète depuis la fin de l’année 2019. Responsable des symptômes associés à la maladie appelée Covid-19, ce virus a complètement bouleversé nos vies et la plupart de nos sociétés.
Un nouveau virus mortel vient de faire son apparition en Chine. A ce jour, plus de 2 000 personnes ont été infectées depuis le mois de décembre et 80 en sont mortes. 3 cas ont été identifiés en France. Quel est ce virus, d’où provient-il et comment se transmet-il ?
Votre abonnement à la lettre d’information : Muséum de Toulouse
En raison d’une panne de climatisation et de la canicule en cours, le Muséum centre-ville est fermé temporairement.
Les visites et activités programmées sont annulées. Nous vous informerons de sa réouverture dès que la situation sera rétablie.
Nous vous prions de nous excuser pour la gêne occasionnée.